Kawasaki Disease
- Overview
- Diagnostic Criteria
- Incomplete Kawasaki Disease
- Coronary Artery Aneurysms
- Treatment
- Echocardiographic Monitoring
- Complications
- Long-Term Outcomes
- Key Research Papers
- Connections
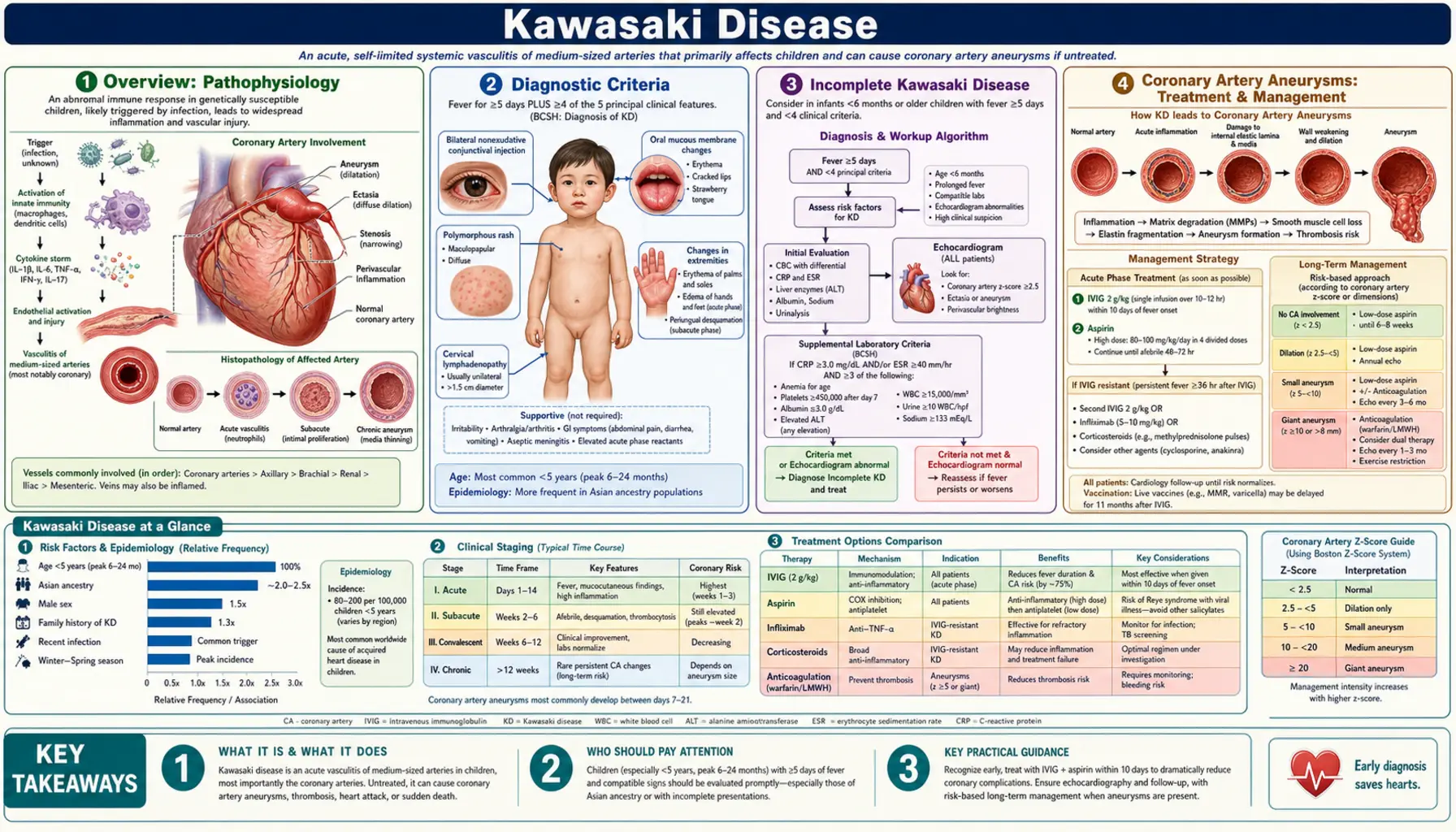
Overview
Kawasaki disease (KD) is a self-limited, acute systemic vasculitis that predominantly affects children under 5 years of age. It is the leading cause of acquired heart disease in children in developed countries, having largely displaced acute rheumatic fever in that role over the past several decades. The condition was first described in Japan in 1967 by pediatrician Tomisaku Kawasaki, who reported 50 children with a distinctive fever-plus-rash-plus-lymphadenopathy illness he called "acute febrile mucocutaneous lymph node syndrome."
KD is classified as a medium-vessel vasculitis. Inflammation targets the walls of medium-caliber arteries throughout the body, but the coronary arteries are the vessels most vulnerable to lasting damage. Unchecked inflammation can cause the coronary arterial wall to weaken and balloon outward, forming coronary artery aneurysms (CAA) — the complication that defines long-term prognosis.
The disease peaks between 6 months and 5 years of age, with a typical patient being a 3-year-old male. Boys are affected roughly 1.5 times more often than girls. Incidence is highest in East Asian children (Japan, Korea, Taiwan), where rates of 200–300 per 100,000 children under 5 years are reported annually. In the United States, incidence is approximately 19–25 per 100,000 children under 5 years, translating to roughly 5,000–6,000 hospitalizations per year. Children of Asian or Pacific Islander descent in the US have substantially higher rates than other ethnic groups.
The underlying cause of KD remains unknown despite more than five decades of research. Leading hypotheses involve an abnormal immune response to one or more ubiquitous infectious triggers (viral or bacterial) in genetically susceptible individuals. No single pathogen has been confirmed. Seasonal clustering (winter-spring peaks) and epidemic waves support an infectious or environmental exposure trigger, but person-to-person transmission has not been demonstrated.
Diagnostic Criteria
Kawasaki disease is a clinical diagnosis — no single laboratory test or imaging finding is pathognomonic. The American Heart Association (AHA) criteria require fever lasting 5 or more days plus at least 4 of the following 5 principal features:
- Bilateral non-purulent (non-exudative) conjunctival injection — redness of the bulbar conjunctivae without discharge; begins within days of fever onset; limbic sparing (a pale ring around the iris) is characteristic.
- Polymorphous rash — appears on the trunk and extremities within the first 5 days; most commonly a maculopapular or morbilliform eruption; may be urticarial, erythema multiforme-like, or scarlatiniform; vesicular or bullous lesions are not consistent with KD.
- Oropharyngeal changes — includes any combination of: strawberry tongue (erythema and prominence of fungiform papillae), lip erythema and cracking/fissuring, and diffuse oropharyngeal erythema. Ulceration, vesicles, and exudate are atypical.
- Changes of the extremities — acute phase: erythema and/or induration (firm swelling) of the palms and soles; subacute/convalescent phase (weeks 2–3): periungual desquamation beginning at the fingertips and spreading proximally. This peeling is essentially pathognomonic when present.
- Cervical lymphadenopathy — at least one lymph node measuring >1.5 cm in diameter; typically unilateral, involves a single anterior cervical node, and is often firm and minimally tender.
A child with fever for 5 or more days plus all 5 features can be diagnosed with classic (complete) KD. In the AHA guidelines, a patient with 4 days of fever can be diagnosed if 4 or more criteria are present and an experienced clinician considers the diagnosis consistent. Fever is typically high (39–40 °C), remittent, and unresponsive to antipyretics and antibiotics — a pattern that itself is a clinical clue.
The fever and most mucocutaneous features resolve within 2–4 weeks without treatment. However, coronary artery inflammation begins early in the course and may progress despite apparent clinical improvement, making early diagnosis and treatment critical.
Incomplete Kawasaki Disease
Incomplete (atypical) Kawasaki disease refers to children who have fever for 5 or more days and only 2 or 3 of the 5 principal features, yet develop coronary artery abnormalities (CAA) consistent with KD. The term "incomplete" is preferred over "atypical" because the missing features likely reflect the same disease process at an earlier or milder stage, not a different disease.
Incomplete KD is particularly dangerous because it is more likely to be missed. Infants younger than 12 months are at highest risk: they frequently present with fever alone or with very few classic features, yet have the highest rates of CAA — up to 25% in some series — if untreated. Any infant under 6 months with unexplained fever lasting 7 or more days warrants echocardiography even without other KD features.
The AHA/American Academy of Pediatrics (AAP) diagnostic algorithm for incomplete KD uses laboratory and echocardiographic criteria when clinical features are insufficient:
- CRP ≥3.0 mg/dL or ESR ≥40 mm/hr in the context of prolonged unexplained fever
- Supplemental laboratory criteria (3 or more of the following support the diagnosis):
- Albumin ≤3.0 g/dL
- Anemia for age
- Elevated alanine aminotransferase (ALT)
- Platelet count >450,000/mm³ after day 7 of fever
- White blood cell count ≥15,000/mm³
- Urinalysis with ≥10 white blood cells per high-power field (sterile pyuria)
- Echocardiographic criteria: Z-score ≥2.5 for the left anterior descending (LAD) or right coronary artery (RCA), or other echocardiographic features of KD (perivascular brightness, lack of tapering, decreased left ventricular function, mitral regurgitation, pericardial effusion)
Per the AHA algorithm, if CRP and ESR are elevated in a child with prolonged fever and 2–3 features, supplemental labs are checked. If 3 or more supplemental criteria are met, treatment should be initiated. If supplemental labs are equivocal but clinical suspicion remains high, echocardiography is performed — and treatment given if echo is abnormal. A normal echo in the first week does not exclude the diagnosis; repeat imaging at 2 weeks is essential.
Coronary Artery Aneurysms
The defining complication of Kawasaki disease is damage to the coronary arteries. Without treatment, approximately 25% of children develop coronary artery aneurysms (CAA). With standard IVIG therapy given within the first 10 days of fever, this rate drops to approximately 4%.
Aneurysms are classified by the internal luminal diameter or, in children, by the coronary Z-score (number of standard deviations above the mean for body surface area):
- Small aneurysm: internal diameter <5 mm (in children <5 years), or Z-score <2.5 in older children
- Medium aneurysm: Z-score 2.5 to <5.0, or diameter 5–8 mm
- Large aneurysm: Z-score 5.0 to <10, or diameter ≥8 mm
- Giant aneurysm: Z-score ≥10, or internal diameter ≥8 mm (some definitions use ≥6 mm in small children)
The proximal left anterior descending (LAD) and right coronary artery (RCA) are most frequently involved. The left main coronary artery (LMCA) is also affected in some cases. Aneurysms are rarely solitary — when present, multiple segments are typically involved.
Small and medium aneurysms often regress on follow-up echocardiography over months to years as the inflammatory process resolves, but regression does not necessarily indicate restoration of normal arterial wall structure. The internal elastic lamina may be permanently disrupted, predisposing to accelerated atherosclerosis, stenosis, and thrombosis decades later.
Giant aneurysms carry the highest risk. Turbulent flow within the dilated segment promotes thrombosis, which can cause acute myocardial infarction — even in toddlers. Children with giant aneurysms require aggressive long-term anticoagulation and cardiology surveillance throughout life. Stenosis developing at the inlet and outlet of a regressing aneurysm is a well-recognized late complication that may precipitate ischemia without obvious thrombosis.
Treatment
The cornerstone of KD treatment is intravenous immunoglobulin (IVIG) combined with aspirin, ideally initiated within the first 10 days of fever onset — and as early as possible once the diagnosis is established.
Primary Treatment
- IVIG 2 g/kg as a single infusion over 10–12 hours. This regimen was established by the landmark 1991 Newburger trial (PMID 1574194) as superior to the prior multi-dose protocol. IVIG reduces fever, systemic inflammation, and coronary artery risk.
- High-dose aspirin 80–100 mg/kg/day in 4 divided doses during the acute febrile phase (anti-inflammatory dosing). Once the child has been afebrile for 48–72 hours, aspirin is stepped down to low-dose 3–5 mg/kg/day once daily (antiplatelet dosing). Low-dose aspirin is continued for at least 6–8 weeks if there are no coronary abnormalities, or indefinitely in children with persistent CAA.
IVIG Resistance
Approximately 10–20% of children do not defervesce within 36 hours after the initial IVIG infusion — a phenomenon called IVIG resistance. These children are at higher risk for CAA and require additional therapy:
- Second dose of IVIG 2 g/kg — most common next step, with approximately 50–60% response rate.
- Infliximab — a TNF-alpha blocker (monoclonal antibody); evidence from the Tremoulet 2014 Lancet trial (PMID 23275434) supports its use as an alternative or adjunct; particularly useful in children who fail a second IVIG dose.
- Corticosteroids — intravenous methylprednisolone or oral prednisolone; used as primary therapy in Japan for high-risk cases per the Kobayashi/Egami/Sano scoring systems, and as rescue therapy in IVIG-resistant disease in Western centers.
- Cyclosporine, anakinra (IL-1 blocker) — emerging options for refractory cases in tertiary centers.
Risk Prediction Scores
Three Japanese scoring systems predict IVIG resistance before treatment is given, allowing risk stratification:
- Kobayashi score (PMID 16950983): 7 variables including sodium, ALT, neutrophil percentage, CRP, platelet count, age, and day of illness; score ≥4 predicts resistance.
- Egami score (PMID 16720205): 5 variables (ALT, CRP, platelet count, age, day of illness); simpler than Kobayashi.
- Sano score (PMID 17719063): 3 variables (AST, neutrophil percentage, sodium); highest positive predictive value in some cohorts.
These scores perform better in Japanese populations than in Western cohorts and should not be used as the sole basis for treatment decisions outside Japan, but they provide useful prognostic context.
Echocardiographic Monitoring
Echocardiography is essential at three standard time points in every child diagnosed with KD:
- At diagnosis (or as soon as possible after) — establishes a baseline and may reveal early coronary changes or pericardial effusion.
- At 2 weeks — peak period for coronary aneurysm formation; the most critical follow-up study.
- At 6–8 weeks — documents resolution or persistence of coronary abnormalities; guides decision on continuing aspirin and scheduling further surveillance.
All coronary artery measurements should be expressed as Z-scores normalized for body surface area, not absolute diameter alone, because coronary artery dimensions scale with body size. The proximal segments of the LAD and RCA are most susceptible to aneurysm formation and must be measured at every study. The LMCA, proximal circumflex, and posterior descending artery are also evaluated.
If the initial and 2-week echocardiograms are normal and the child had classic KD with good clinical response to IVIG, no further echocardiographic follow-up is typically required beyond the 6–8 week study. If coronary abnormalities are detected, the frequency and duration of surveillance depend on the risk tier defined in the 2017 AHA guidelines:
- Tier I (no coronary changes): echo at 2 weeks and 6–8 weeks; no long-term cardiology follow-up required.
- Tier II (transient dilation only, normalized by 4–6 weeks): echo at 2 weeks, 4–6 weeks, and 12 months.
- Tier III (small persistent aneurysm): annual echo; cardiology follow-up; aspirin until regression confirmed.
- Tiers IV and V (medium to giant aneurysm): frequent echo (every 3–6 months), anticoagulation, stress testing, possibly coronary angiography, lifelong cardiology care.
Complications
While coronary artery aneurysms are the most feared complication, KD produces systemic vasculitis that can affect multiple organ systems:
- Myocarditis — present in the acute phase in most children; usually subclinical but can cause reduced left ventricular ejection fraction, tachycardia out of proportion to fever, and gallop rhythm; detectable by cardiac MRI even when echo is normal.
- Pericarditis / pericardial effusion — small effusions are common acutely; rarely hemodynamically significant.
- Mitral regurgitation — mild MR is common acutely due to papillary muscle inflammation; usually resolves with treatment.
- Hydrops of the gallbladder — painless, non-tender right upper quadrant mass; resolves with IVIG; important to distinguish from surgical abdomen.
- Arthritis / arthralgia — affects approximately one-third of children; large joints in the acute phase, small joints in the subacute phase.
- Aseptic meningitis — CSF pleocytosis with normal protein and glucose; children are often irritable out of proportion to other findings.
- Urethritis / meatitis — sterile urethral inflammation producing dysuria and periurethral erythema; sterile pyuria on urinalysis is common.
- Macrophage Activation Syndrome (MAS) — rare but life-threatening hyperinflammatory complication; presents with worsening cytopenias, coagulopathy, and markedly elevated ferritin; treated with high-dose steroids and cyclosporine.
- Sensorineural hearing loss — reported in small case series; thought to be due to cochlear vasculitis.
Long-Term Outcomes
The great majority of children who receive timely treatment with IVIG recover completely with no lasting cardiac sequelae. Fever resolves, inflammatory markers normalize, and mucocutaneous features fade over 2–4 weeks. For children who never develop coronary abnormalities, long-term prognosis is essentially normal.
However, for the minority with persistent or giant aneurysms, KD becomes a lifelong cardiovascular disease. Giant aneurysms do not fully regress and carry compounding risks over decades:
- Thrombotic occlusion causing acute MI (can occur in infancy or decades later)
- Stenosis at aneurysm inlet/outlet as fibrous tissue accumulates
- Accelerated atherosclerosis within the abnormal arterial wall segment
Adults who had KD in childhood — many of whom were never diagnosed — may present with premature coronary artery disease, MI, or sudden cardiac death. This has led to retrospective recognition of KD as a cause of "unexplained" MI in young adults, particularly those of Asian descent.
Multisystem Inflammatory Syndrome in Children (MIS-C), identified during the COVID-19 pandemic, shares several features with KD — prolonged fever, conjunctivitis, rash, mucocutaneous changes, and coronary artery dilation — and has been called a "Kawasaki-like" illness. MIS-C typically affects older children (median age 8–9 years), more often causes gastrointestinal symptoms, shock, and myocarditis, and follows SARS-CoV-2 infection by 2–6 weeks. The two conditions are clinically distinct but may share immunopathogenic mechanisms (PMID 32360583).
Long-term management for high-risk KD patients includes aspirin (and anticoagulation for giant aneurysms), exercise restriction proportional to aneurysm size, periodic stress testing and coronary CT or MR angiography, and statin therapy in some guidelines. Percutaneous coronary intervention and bypass grafting are occasionally required for stenotic lesions.
Key Research Papers
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
PubMed Topic Searches
- Kawasaki disease coronary artery aneurysm
- Kawasaki disease IVIG treatment
- Incomplete Kawasaki disease diagnosis
- Kawasaki disease IVIG resistance infliximab
- MIS-C Kawasaki-like COVID-19
Connections
- Pediatrics
- Intussusception
- Croup
- Juvenile Idiopathic Arthritis
- Cardiology — Acquired Heart Disease
- Rheumatology — Vasculitis
- Infectious Disease — Fever of Unknown Origin