Biliary Atresia
- What Is Biliary Atresia?
- Types of Biliary Atresia
- Pathogenesis
- Clinical Presentation
- Diagnosis
- Kasai Hepatoportoenterostomy
- Liver Transplantation
- Prognosis and Long-Term Outcomes
- Key Research Papers
- Connections
- Featured Videos
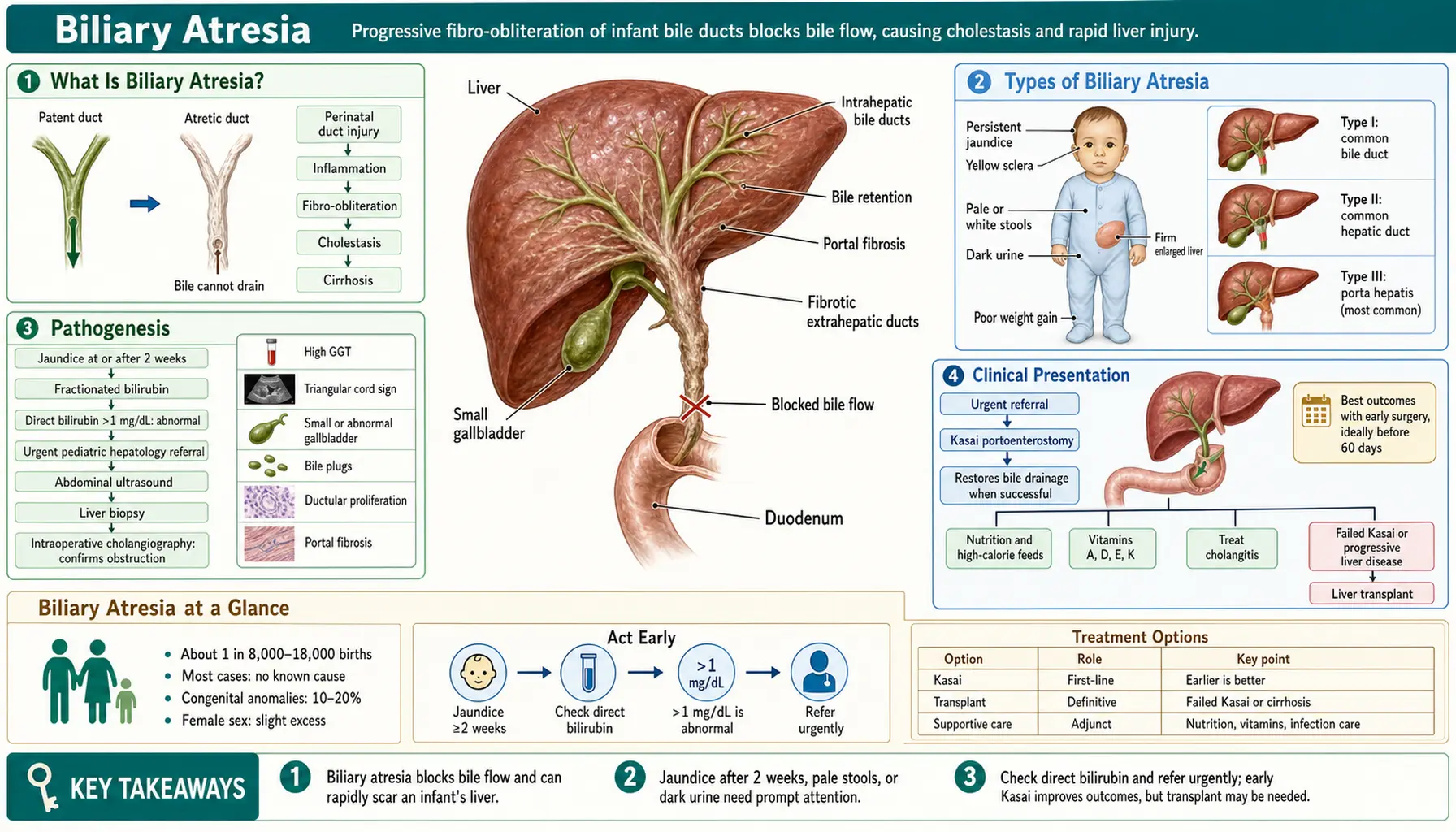
What Is Biliary Atresia?
Biliary atresia (BA) is a progressive fibro-obliterative cholangiopathy of infancy in which the extrahepatic bile ducts become inflamed, scarred, and obliterated, preventing bile from draining from the liver into the intestine. It is the most common cause of neonatal cholestasis requiring surgical intervention and the leading indication for pediatric liver transplantation worldwide, accounting for approximately 40–50% of all pediatric liver transplants.
The disease is not simply a congenital malformation present at birth in a static sense. In the most common form, the bile ducts may be initially patent at birth and only subsequently undergo progressive inflammatory destruction. This dynamic, ongoing process distinguishes biliary atresia from structural ductal anomalies like choledochal cyst and makes early diagnosis and timing of surgery critically important.
Incidence: Biliary atresia affects approximately 1 in 10,000–15,000 live births in Western populations. Incidence is higher in East Asian populations (1:6,000–1:8,000 in Taiwan and Japan). In the United States, approximately 300–400 new cases are diagnosed each year. There is a slight female predominance (female-to-male ratio approximately 1.1–1.4:1), the reason for which is unknown.
Etiology: The precise cause of biliary atresia remains incompletely understood. Leading hypotheses include:
- Viral triggers: Perinatal viral infections — particularly cytomegalovirus (CMV) and rotavirus — have been most strongly implicated. CMV IgM seropositivity is found in 30–40% of BA infants in some series. Animal models using rhesus rotavirus injection in newborn mice reproducibly induce a BA-like phenotype, suggesting viral-triggered immune activation of bile duct epithelium.
- Immune/inflammatory dysregulation: Even in cases without demonstrable viral trigger, there is evidence of aberrant innate and adaptive immune activation targeting cholangiocytes (bile duct cells). Elevated Th1 cytokines (IFN-γ, TNF-α), natural killer cell infiltration, and regulatory T cell defects have all been described in BA liver tissue.
- Developmental abnormality: The embryonic/fetal form of BA (present from birth with additional structural anomalies) suggests that disrupted morphogenesis of the biliary system during organogenesis plays a role in a subset of patients.
Importantly, biliary atresia is not hereditary — it does not follow Mendelian inheritance patterns, and the risk to siblings is not meaningfully elevated above the population rate.
Types of Biliary Atresia
Biliary atresia is not a single uniform entity. The two principal clinical forms differ in their timing of onset, associated anomalies, and (in some evidence) surgical outcomes.
Perinatal Form (Acquired / Isolated BA)
The perinatal form accounts for 65–90% of all biliary atresia cases. In this form, bile ducts are believed to be initially patent at or shortly after birth. The characteristic clinical course is:
- The normal physiological jaundice of the newborn (indirect hyperbilirubinemia, unconjugated) appears to resolve in the first 1–2 weeks, as expected.
- Then, as the inflammatory and fibrosing process obliterates the extrahepatic ducts, jaundice reappears or persists in weeks 2–4 of life, now with a direct/conjugated hyperbilirubinemia component — a finding that is always pathological and mandates immediate workup.
- No other structural anomalies are present (isolated BA).
This clinical window of apparent improvement followed by re-emergence is a key reason why BA can be diagnosed later than optimal: it mimics normal neonatal jaundice resolution in its early phase. The stool color card programs implemented in Taiwan and Japan, where caregivers assess infant stool color at weeks 1 and 1 month, have successfully shifted diagnosis earlier.
Embryonic/Fetal Form (Syndromic BA)
The embryonic/fetal form accounts for 10–35% of cases (estimates vary). In this form:
- Jaundice is persistent from birth — there is no window of apparent resolution, because the ductal abnormality is present from the start of extrauterine life.
- It is associated with additional structural congenital anomalies in approximately 10–20% of all BA patients — a cluster called BASM syndrome (Biliary Atresia Splenic Malformation).
BASM syndrome (also called the "splenic malformation syndrome") includes one or more of:
- Polysplenia (multiple small spleens or a bilobed spleen) — most characteristic finding; present in ~90% of syndromic BA
- Cardiac defects — most commonly interrupted inferior vena cava with azygos continuation, atrial septal defects, or more complex lesions
- Situs inversus (reversal of left-right organ positioning) or situs ambiguus (heterotaxy)
- Intestinal malrotation, preduodenal portal vein, absence of the hepatic artery, or other vascular anomalies
The syndromic form is thought to reflect a defect in laterality determination during early embryogenesis — the same developmental pathway that establishes left-right asymmetry also influences biliary morphogenesis. Genes involved in left-right axis specification (CFC1, CRELD1, and others) have been implicated in some BASM patients. Importantly, some studies suggest that the BASM/syndromic form may have poorer Kasai outcomes than isolated BA, though results are not uniform across series.
Pathogenesis
The fundamental pathological process in biliary atresia is a progressive inflammatory fibrosis of the extrahepatic bile ducts that extends intrahepatic if untreated. Understanding this sequence explains both why early surgery is so important and why liver transplantation remains necessary for most patients even after successful Kasai surgery.
Step 1 — Bile duct inflammation: A triggering event (viral infection, immune dysregulation, or developmental insult) initiates inflammatory infiltration of the extrahepatic bile duct wall. Cholangiocytes respond by upregulating adhesion molecules and inflammatory cytokines, attracting NK cells, macrophages, and activated CD4+ T cells. The bile duct epithelium begins to sustain injury.
Step 2 — Progressive fibrous obliteration: Ongoing inflammation drives fibrous replacement of the bile duct wall, progressively narrowing and then obliterating the ductal lumen. This process begins in the extrahepatic ducts — the common bile duct, hepatic ducts, and cystic duct — and progresses both distally (toward the duodenum) and proximally (toward the liver hilum). By the time of surgical exploration, in most patients no residual ductal lumen is identifiable; the ducts have been replaced by fibrous tissue.
Step 3 — Biliary obstruction: Complete obliteration of the extrahepatic ducts causes total biliary obstruction. Bile, which is produced continuously by hepatocytes, can no longer drain into the intestine. It backs up into the liver, causing:
- Cholestatic jaundice — conjugated (direct) bilirubin accumulates in blood
- Acholic (pale) stools — absence of bilirubin pigments reaching the intestine
- Dark urine — conjugated bilirubin excreted by the kidney
- Fat malabsorption — absence of bile salts in the intestine impairs fat and fat-soluble vitamin (A, D, E, K) absorption
Step 4 — Progressive biliary cirrhosis: Bile acid retention is directly toxic to hepatocytes. Bile acids accumulate intracellularly, activate apoptotic pathways, and promote reactive oxygen species generation. Simultaneously, portal tract fibrosis expands, bile duct proliferation occurs (a reactive response to obstruction), and the hepatic architecture is progressively distorted. Within weeks to months, biliary cirrhosis develops.
Step 5 — Liver failure: If biliary obstruction is not relieved — either by successful Kasai surgery or liver transplantation — progressive biliary cirrhosis leads to end-stage liver failure with portal hypertension, synthetic dysfunction (coagulopathy, hypoalbuminemia), and ultimately death, typically by age 2 years. This is the universal natural history of untreated biliary atresia.
Clinical Presentation
Biliary atresia presents in the neonatal and early infant period. The baby typically appears well at birth with normal birth weight and early feeding — the disease is not apparent immediately at delivery. The key clinical signs emerge over the first weeks of life.
Prolonged Jaundice
The cardinal presenting feature is persistent or worsening jaundice beyond 14 days of age. In contrast to physiological neonatal jaundice (predominantly unconjugated, peaks at days 3–5, resolves by 2 weeks in term infants), the jaundice of biliary atresia is due to conjugated (direct) hyperbilirubinemia.
A direct bilirubin above 1 mg/dL, or more than 20% of the total bilirubin, is always pathological in a neonate — there is no normal level of direct hyperbilirubinemia in a newborn. This is the critical lab threshold that triggers the biliary atresia workup. Any jaundiced infant past 14 days old must have a fractionated bilirubin measured — the color of the baby alone cannot distinguish direct from indirect jaundice.
Acholic (Pale/White) Stools
Pale or acholic stools are the most specific clinical sign of biliary obstruction. Normally, conjugated bilirubin is secreted into bile and converted by intestinal bacteria to urobilinogen and stercobilin, which give stool its brown-yellow color. When bile cannot reach the intestine, stool loses its pigment and becomes pale grey, white, or clay-colored.
Acholic stools are present in up to 80–90% of BA patients at diagnosis. Parents may notice this change but not report it unless specifically asked — pediatricians should ask about stool color at every well-child visit in the first month of life, and stool color cards (now used in Japan, Taiwan, and some European countries) show parents what normal vs. abnormal stool colors look like.
Dark Urine
Tea-colored or dark-yellow urine reflects conjugated bilirubin excreted by the kidneys (conjugated bilirubin, unlike unconjugated bilirubin bound to albumin, is water-soluble and appears in urine). This finding, combined with pale stools, is a classic combination pointing to obstructive jaundice.
Hepatomegaly
The liver enlarges progressively as biliary obstruction and inflammation cause hepatocyte injury and hepatic fibrosis. A firm or hard liver edge on palpation suggests established fibrosis and carries prognostic significance — excessive fibrosis at the time of Kasai surgery correlates with worse long-term outcome. Splenomegaly develops later as portal hypertension supervenes.
Fat-Soluble Vitamin Deficiency
Since bile acids are absent from the intestinal lumen, fat and fat-soluble vitamins (A, D, E, K) are malabsorbed. Early consequences include:
- Coagulopathy — Vitamin K deficiency → prolonged PT/INR (must be distinguished from synthetic liver failure)
- Rickets — Vitamin D deficiency
- Growth failure — fat malabsorption reduces caloric intake; patients need supplemental fat-soluble vitamins and high-calorie formula
Infants with BA often appear otherwise well in the first few weeks despite severe cholestasis — they feed, gain weight modestly, and seem alert. This can falsely reassure caregivers and delay referral. The jaundice and pale stools are the critical clues.
Diagnosis
The diagnosis of biliary atresia requires a systematic workup to exclude other causes of neonatal cholestasis and to confirm the diagnosis before surgical intervention. Time is critical — outcomes deteriorate sharply with delayed surgery.
Initial Laboratory Evaluation
Any jaundiced infant over 14 days old should have a fractionated bilirubin measured. If direct bilirubin exceeds 1 mg/dL or 20% of total, the following are obtained:
- Liver function tests (ALT, AST, GGT, alkaline phosphatase) — typically elevated; GGT is markedly elevated in BA (often >300 U/L), which helps distinguish BA from progressive familial intrahepatic cholestasis (PFIC) syndromes where GGT is normal or low
- Coagulation studies (PT/INR, PTT) — Vitamin K deficiency or synthetic dysfunction
- Complete blood count, albumin, glucose
- Thyroid function (hypothyroidism causes neonatal cholestasis)
- CMV IgM/IgG serology and urine CMV PCR
- Alpha-1-antitrypsin level and phenotype (A1AT deficiency is a key mimic)
- Urine organic acids, plasma amino acids, galactose-1-phosphate (metabolic screen)
- TORCH serologies, hepatitis B surface antigen
Liver Ultrasound
Abdominal ultrasound is performed early in the workup. Key findings in biliary atresia:
- Absent or abnormal gallbladder — the gallbladder is absent or appears as a small, shrunken, atretic structure in 70–90% of BA cases. A normal-appearing gallbladder on ultrasound makes BA less likely but does not exclude it.
- Triangular cord sign — echogenic density at the hepatic hilum representing fibrous tissue obliterating the portal plate; has a positive predictive value of up to 90% when present (though sonographer experience matters)
- Normal intrahepatic bile ducts (the intrahepatic ducts are not dilated in BA, distinguishing it from choledochal cyst)
- Liver echogenicity may be increased; assess for spleen and portal vein for evidence of portal hypertension
Hepatobiliary Scintigraphy (HIDA Scan)
A technetium-99m iminodiacetic acid (HIDA) scan assesses the hepatic uptake of a radiotracer and its excretion into the biliary system and intestine. In biliary atresia:
- Hepatocyte uptake is intact (the tracer concentrates in the liver normally) — reflecting preserved hepatocellular function early in the disease
- Intestinal excretion is absent — no radiotracer appears in the intestine on delayed imaging at 24 hours
A HIDA scan showing absent intestinal excretion has high sensitivity (~99%) for biliary obstruction, but the specificity for distinguishing BA from severe intrahepatic cholestasis (which also impairs excretion) is limited. Phenobarbital pretreatment (5 mg/kg/day × 5 days) maximizes specificity by inducing bile flow in intrahepatic causes. The HIDA scan is used as a screening tool; it cannot confirm BA alone — it is one piece of the diagnostic mosaic.
Liver Biopsy
Percutaneous liver biopsy is the most important non-surgical diagnostic tool. Classic histological features of biliary atresia include:
- Bile duct proliferation — increased number of small bile ductules in the portal tracts (a reactive response to obstruction)
- Bile plugs — inspissated bile in dilated ductules
- Portal tract fibrosis — varying degrees of portal-portal and portal-central fibrosis; the degree of fibrosis at biopsy has prognostic value
- Lobular cholestasis — canalicular and hepatocellular bile stasis
Biopsy has a reported sensitivity of 90–95% and specificity of 60–80% for BA. It is most useful for excluding other diagnoses (A1AT deficiency has characteristic PAS-positive, diastase-resistant globules; PFIC has specific ultrastructural patterns; neonatal hepatitis shows a distinctive giant-cell hepatitis pattern).
Intraoperative Cholangiography — Gold Standard
The definitive diagnosis of biliary atresia is made at intraoperative cholangiography. Under general anesthesia, the gallbladder (if present) or a duct remnant is cannulated and contrast is injected. In biliary atresia:
- No opacification of the extrahepatic bile ducts or duodenum — contrast is injected but cannot flow due to fibrous obliteration
- The hepatic hilum cannot be traversed by contrast
When intraoperative cholangiography confirms BA, the surgical team immediately proceeds to Kasai hepatoportoenterostomy — the definitive surgical treatment — without closing and re-operating later. This is why the diagnostic and surgical teams must be prepared for immediate intervention at the time of cholangiogram.
Kasai Hepatoportoenterostomy
The Kasai hepatoportoenterostomy — named for Japanese surgeon Morio Kasai, who first described the procedure in 1959 — is the primary surgical treatment for biliary atresia and the only intervention capable of restoring bile flow using the infant's own anatomy, potentially deferring or (in a minority of cases) obviating the need for liver transplantation.
Surgical Technique
The Kasai procedure is a Roux-en-Y hepatoportoenterostomy:
- The atretic extrahepatic bile ducts and gallbladder (en bloc) are dissected and excised up to their junction at the hepatic hilum — the fibrous cone at the porta hepatis.
- The liver hilum is exposed and the fibrous tissue is excised flush with the liver plate, exposing the cut surface of the portal plate where microscopic bile ductules communicate with the intrahepatic biliary system.
- A segment of jejunum is brought up as a Roux limb (typically 40–45 cm) and anastomosed directly to the transected portal plate — the intestine is sewn to the liver hilum, bypassing the absent extrahepatic ducts entirely.
- Bile drains directly from the microscopic ductules at the cut hepatic hilum into the Roux limb and onward to the intestine.
The procedure is typically performed open, though laparoscopic Kasai is performed at specialized centers with comparable short-term outcomes but longer operative times.
Age at Surgery — The Critical Variable
The most powerful predictor of Kasai success is age at time of surgery:
- Under 45 days: Highest success rates — approximately 80% achieve initial bile drainage. Fibrosis is less advanced; bile ductules at the hepatic hilum are more patent and better preserved.
- 45–60 days: Good outcomes; 60–80% achieve bile drainage. Most published guidelines target this window as the acceptable maximum for non-emergency referral.
- 60–90 days: Outcomes decline significantly — approximately 40–50% achieve adequate bile flow. Native liver survival at 5 years falls substantially.
- Over 90 days: Poor surgical outcome; fibrosis at the hilum is typically advanced; less than 20–30% achieve adequate bile drainage. At this stage, primary liver transplantation listing may be preferable at experienced centers, though Kasai is still offered.
The 60-day threshold is a frequently cited clinical rule: every week of delay from 4–12 weeks of age reduces the probability of adequate bile drainage. This drives the urgency of early diagnosis — in countries with stool card screening programs, median age at Kasai is 50–55 days vs. 60–70 days in countries without systematic screening.
Outcomes After Kasai Surgery
Success of the Kasai operation is defined as restoration of adequate bile drainage, typically measured as total serum bilirubin falling below 2 mg/dL within 3–6 months of surgery (the "jaundice clearance" endpoint).
- Approximately 50% of successfully operated patients achieve jaundice clearance in Western series; up to 70% in high-volume Japanese centers with early referral.
- Among patients who achieve jaundice clearance, approximately 50% retain their native liver at 5 years and a meaningful fraction maintain native liver function into adulthood.
- Among patients who do not achieve jaundice clearance after Kasai, most will require liver transplantation within 1–2 years.
- Overall, across all BA patients regardless of Kasai outcome, approximately 80% will require liver transplantation by adulthood.
Post-Kasai Cholangitis
Ascending cholangitis is the most common serious complication after Kasai, occurring in 40–60% of patients in the first year. The Roux limb lacks normal intestinal flora gating and allows bacterial ascent into the bile drainage pathway. Cholangitis presents as:
- Fever (>38.5°C) in an infant who had achieved bile drainage
- Sudden return of jaundice (rising bilirubin)
- Acholic stools returning
- Elevated inflammatory markers (CRP, WBC)
Each episode of cholangitis can damage the intrahepatic bile ducts and accelerate the progression of hepatic fibrosis, even after successful Kasai. Prophylactic antibiotics (e.g., trimethoprim-sulfamethoxazole) are used at most centers post-Kasai, though evidence for prophylactic benefit is mixed. Cholangitis episodes require prompt IV antibiotic treatment.
Liver Transplantation
Liver transplantation is the definitive curative treatment for biliary atresia when the Kasai procedure fails to restore adequate bile drainage, or when progressive cirrhosis and liver failure supervene despite initial Kasai success. Without any treatment, infants with biliary atresia die from liver failure by approximately age 2 years — transplantation is life-saving in this context.
Indications for Transplant After Kasai
- Failed bile drainage — persistent severe jaundice (bilirubin >6 mg/dL) at 3–6 months post-Kasai despite intact surgery
- Decompensated cirrhosis — ascites, variceal bleeding, synthetic failure (INR >1.5 despite Vitamin K, albumin <2.5 g/dL) despite adequate bile drainage
- Recurrent severe cholangitis — repeated infections accelerating liver damage
- Hepatopulmonary syndrome or portopulmonary hypertension
- Failure to thrive refractory to nutritional support
- Hepatocellular carcinoma (rare but reported in BA with cirrhosis)
Timing and Organ Source
Because BA infants requiring transplant are small (often <10 kg), cadaveric whole-organ transplant requires size-matched donors — supply is severely limited for infants. Techniques that have expanded donor availability include:
- Reduced-size liver transplantation — adult or older child cadaveric donor liver surgically reduced to fit an infant recipient
- Split liver transplantation — one adult cadaveric liver divided to provide a left lateral segment (segments II-III) for a small child and the right lobe for an adult, maximizing one donor organ for two recipients
- Living-donor liver transplantation (LDLT) — a parent or relative donates their left lateral segment; LDLT accounts for up to 80% of pediatric liver transplants in Japan and has expanded to many Western centers; it allows elective timing and avoids wait-list mortality
Outcomes with Transplant
Pediatric liver transplantation for biliary atresia has excellent outcomes at experienced centers:
- 1-year patient survival: 90–95%
- 5-year patient survival: 85–90%
- 10-year patient survival: 80–85%
Children who undergo successful liver transplantation for BA typically enjoy normal growth, development, and quality of life with chronic immunosuppression (usually tacrolimus ± mycophenolate). The goal is minimizing immunosuppression over time to reduce the risks of opportunistic infections, post-transplant lymphoproliferative disorder (PTLD), and nephrotoxicity while preventing rejection.
Unlike in adults, where BA does not recur in the transplanted liver (BA is not a systemic disease — it targets only the patient's own bile ducts), children with BA and transplant have excellent long-term graft survival. The main long-term concerns are chronic kidney disease (tacrolimus nephrotoxicity) and management of adolescent non-adherence to immunosuppression, which is a leading cause of late graft loss in pediatric transplant recipients.
Prognosis and Long-Term Outcomes
The prognosis of biliary atresia has improved dramatically over the past 50 years with the widespread adoption of the Kasai procedure and advances in pediatric liver transplantation. Outcomes are highly variable and depend on several interrelated factors.
Factors Affecting Native Liver Survival After Kasai
- Age at Kasai surgery — the single most important modifiable factor; early surgery (<45–60 days) is the highest-impact intervention
- Jaundice clearance — achieving serum bilirubin <2 mg/dL by 3–6 months post-Kasai is the strongest predictor of long-term native liver survival
- Degree of hepatic fibrosis at time of surgery — more advanced fibrosis (Metavir F3-F4 at Kasai) predicts worse outcomes regardless of surgical success
- Surgical volume and experience — Kasai outcomes are significantly better at high-volume centers (≥5 Kasai procedures per year) than at low-volume centers; this is one of the strongest volume-outcome relationships in pediatric surgery
- Number and severity of post-Kasai cholangitis episodes
- CMV status — some studies show worse outcomes in CMV-seropositive infants, though this is not uniformly reproduced
- BASM/syndromic form — some series report worse Kasai outcomes in the syndromic form
Long-Term Native Liver Survival Statistics
Published long-term data from high-volume centers show:
- 5-year native liver survival: 40–55% across all patients (higher in those achieving early jaundice clearance)
- 10-year native liver survival: 25–40%
- 20-year native liver survival: 20–25% (a meaningful minority of patients with excellent early response carry their native liver well into adulthood)
Quality of Life and Adult Outcomes
Patients who carry their native liver long-term face ongoing surveillance for:
- Progressive portal hypertension — esophageal varices, hypersplenism, and ascites can develop even in patients with apparent bile drainage clearance if cirrhosis was established before Kasai
- Fat-soluble vitamin deficiency — requires lifelong monitoring and supplementation of vitamins A, D, E, K
- Bone disease (metabolic bone disease from Vitamin D deficiency and cholestasis-related effects on bone metabolism)
- Hepatocellular carcinoma screening — rare but reported in older BA survivors with cirrhosis; annual AFP + ultrasound recommended
- Transition to adult hepatology care — essential and often challenging; BA patients reaching adulthood require gastroenterology/hepatology teams experienced with this population
Overall, patients with biliary atresia and liver transplantation lead essentially normal lives with appropriate medical monitoring. Children who received transplants in infancy are today reaching adulthood, attending university, working, and in some cases having children of their own. The psychosocial burden of chronic illness and lifelong immunosuppression is real but manageable.
Key Research Papers
- Search PubMed PMID: 15488218 — Authoritative overview of pathogenesis, diagnosis, and surgical management of biliary atresia, discussing both the perinatal and embryonic forms and the role of the Kasai procedure.
- Search PubMed PMID: 22418886 — Comprehensive research agenda summary covering BA immune pathogenesis, genetic factors, and emerging therapeutic targets including anti-fibrotic and anti-inflammatory strategies.
- Search PubMed PMID: 21496560 — Multicenter US study (the Childhood Liver Disease Research Network) documenting that anatomic type at Kasai and early jaundice clearance are the strongest predictors of transplant-free native liver survival.
- Search PubMed PMID: 17671195 — NIH workshop report synthesizing evidence on screening strategies for early BA detection, including stool color card programs, and their impact on timing of Kasai surgery and outcomes.
- Search PubMed PMID: 23331918 — National registry study reporting patient and graft survival data from the US multicenter experience, showing excellent long-term outcomes for BA patients receiving liver transplants.
- Search PubMed PMID: 10023497 — Landmark French national cohort study providing population-based data on survival, Kasai success rates, and transplant outcomes, helping establish the modern two-stage (Kasai → transplant) treatment paradigm.
- Search PubMed PMID: 22177990 — Long-term follow-up study characterizing the hepatic and extrahepatic health status of BA patients who survive to adulthood on their native liver, including portal hypertension burden and transplant-free survival determinants.
- Search PubMed PMID: 18801757 — Population-based incidence study documenting BA epidemiology, seasonal clustering, and geographic variation in England and Wales, contributing to evidence of viral etiology.
- Search PubMed PMID: 19307120 — Updated comprehensive review covering etiology, pathogenesis (including immune mechanisms), surgical management, and outcomes of biliary atresia; a widely used clinical reference.
- Search PubMed PMID: 15187805 — North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN) clinical practice guideline defining the diagnostic algorithm for neonatal cholestasis and the threshold for BA workup.
- Murase N et al. Analysis of biliary atresia cases with successful Kasai surgery. Surg Today. 2018. — Search PubMed for age-at-surgery outcome analyses; multiple studies confirm the exponential decline in Kasai success rates beyond 60–90 days of age.
- Shanmugam NP et al. Revisiting the relevance of biliary atresia. Dig Liver Dis. 2009. — PubMed search for post-Kasai cholangitis management; ascending cholangitis remains the dominant early complication driving native liver deterioration after successful surgery.
Additional PubMed searches:
Biliary atresia Kasai hepatoportoenterostomy outcomes Biliary atresia pediatric liver transplant Neonatal cholestasis biliary atresia diagnosis
Connections
- Pediatrics
- Neonatal Jaundice
- Congenital Heart Disease
- Gastroenterology
- Liver Disease
- Endocrinology
- Lab Tests
- Vitamin D
- Jaundice — the general symptom; conjugated hyperbilirubinemia persisting past 14 days is what separates biliary atresia from the physiological kind.
- Vitamin K