Renal Tubular Acidosis
Renal tubular acidosis (RTA) is a group of disorders in which the kidney's tubules fail to properly regulate acid-base balance, producing a characteristic hyperchloremic, non-anion-gap metabolic acidosis despite relatively preserved glomerular filtration. Each type reflects a distinct tubular segment defect and carries its own pattern of serum potassium, urinary findings, and clinical complications.
Interactive Visualization The Sodium–Potassium Pump — why you have a voltage Run the pump that gives every cell its charge — three sodium out, two potassium in, on Mg-ATP — then drop the magnesium and see exactly why low potassium refuses to correct. Launch →Table of Contents
- Overview and Classification
- Type 1: Distal RTA (dRTA)
- Type 2: Proximal RTA and Fanconi Syndrome
- Type 4: Hyperkalemic RTA
- Diagnosis and Laboratory Findings
- Differential Diagnosis
- Treatment and Management
- Prognosis and Long-Term Outcomes
- Key Research Papers
- Connections
- Featured Videos
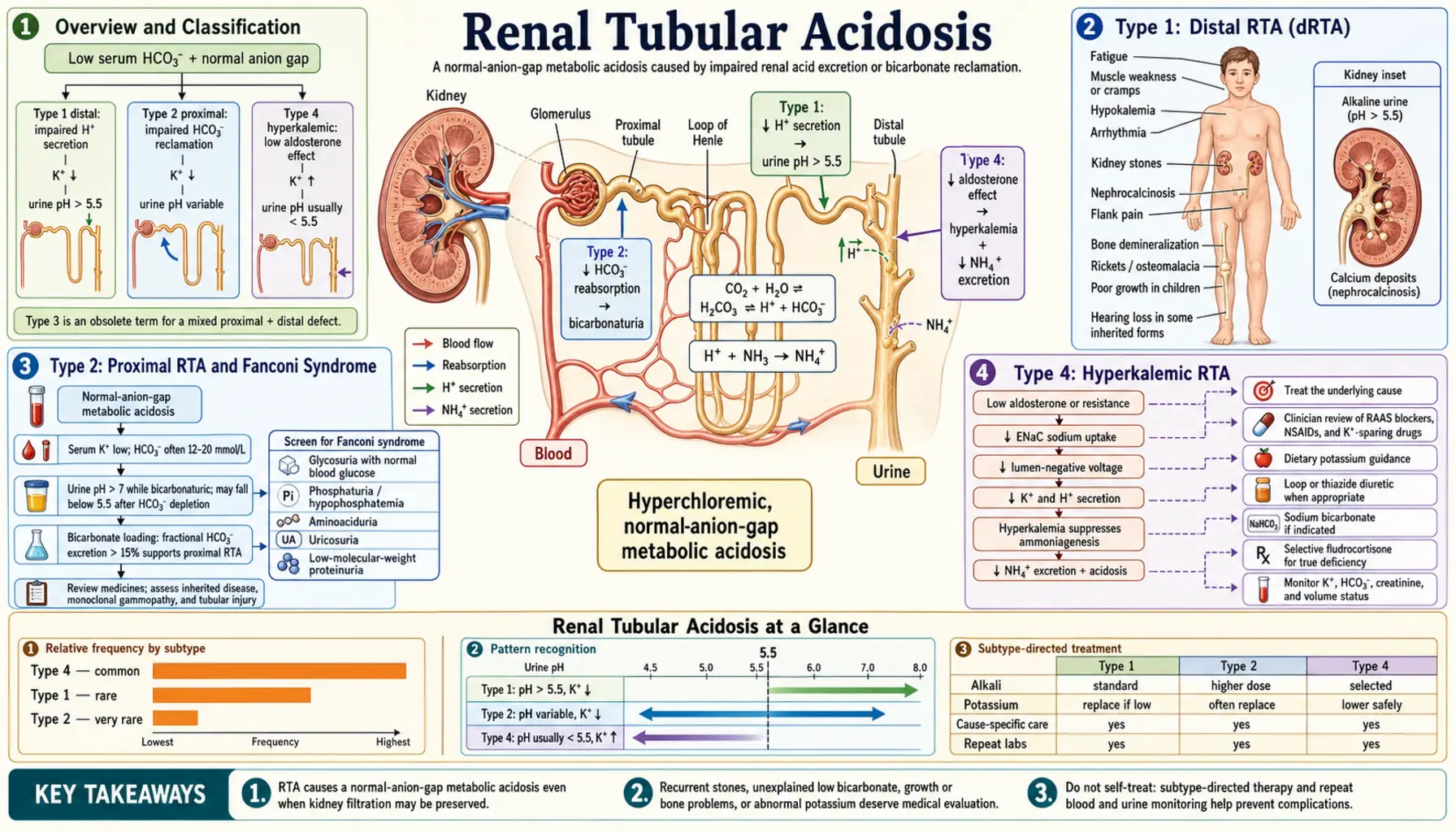
Overview and Classification
Under normal conditions the kidney excretes approximately 1 mEq/kg/day of non-volatile acid generated by protein catabolism. Two tubular processes accomplish this: the proximal tubule reabsorbs the bulk of filtered bicarbonate (80–90%), while the distal collecting duct secretes free protons to generate titratable acid and ammonium. Failure at either site — or an impaired hormonal signal that drives distal acid secretion — produces metabolic acidosis with a normal anion gap (hyperchloremic metabolic acidosis), distinguishing RTA from conditions such as lactic acidosis or diabetic ketoacidosis where an unmeasured anion accumulates.
RTA is numbered by convention rather than discovery order, and Type 3 (a transient carbonic-anhydrase-deficient form seen in infants) has been largely abandoned as a distinct category. The three clinically relevant types are:
- Type 1 (Distal RTA): Collecting duct cannot acidify urine; urine pH persistently above 5.5; hypokalemia; nephrocalcinosis and nephrolithiasis are hallmarks.
- Type 2 (Proximal RTA): Proximal tubule cannot reabsorb bicarbonate; HCO3 spills in urine until serum falls to a new, lower threshold; often accompanied by global proximal tubule dysfunction (Fanconi syndrome).
- Type 4 (Hyperkalemic RTA): Aldosterone deficiency or resistance impairs both potassium and hydrogen excretion in the collecting duct; the most common type in clinical practice, typically occurring in elderly patients with diabetic nephropathy.
Type 1: Distal RTA (dRTA)
Type 1 (distal) RTA results from failure of the alpha-intercalated cells of the cortical and medullary collecting duct to secrete hydrogen ions. The H+-ATPase (vacuolar-type proton pump) on the apical membrane or the anion exchanger AE1 (encoded by SLC4A1) on the basolateral membrane is dysfunctional, either from genetic mutation or acquired immune damage. Because the collecting duct cannot generate a steep H+ gradient, urine pH cannot fall below 5.5 even when systemic acidosis is severe — the classic and diagnostic hallmark of Type 1 RTA.
The ensuing acid retention is buffered partly by skeletal calcium carbonate, releasing calcium into the circulation and urine. This hypercalciuria, combined with the obligatory alkaline urine (which reduces calcium solubility), promotes calcium phosphate crystal precipitation in the renal medulla — nephrocalcinosis — and in the collecting system — nephrolithiasis. Calcium phosphate (brushite) stones, rather than the calcium oxalate stones typical of idiopathic hypercalciuria, are the characteristic stone type. Citrate, normally a stone-inhibitor excreted by tubular cells, is consumed in buffering the systemic acidosis, further removing protection against stone formation.
Potassium handling is disrupted because the failure to secrete H+ redirects the collecting duct to excrete K+ via principal cells in an attempt to maintain electroneutrality, producing hypokalemia. Chronic acidosis in untreated children impairs growth hormone signaling, causing growth retardation.
Causes of acquired dRTA:
- Sjogren syndrome — most common secondary cause; autoantibodies against carbonic anhydrase II or H+-ATPase intercalated cells; RTA may precede sicca symptoms by years
- Systemic lupus erythematosus (SLE) — immune complex deposition in tubular basement membranes
- Primary hyperparathyroidism — hypercalcemia directly impairs collecting duct acidification
- Medullary sponge kidney — cystic dilatation of collecting ducts impairs normal acidification
- Wilson's disease — copper deposition in tubular cells
- Chronic active hepatitis and primary biliary cholangitis
- Amphotericin B — creates pores in tubular cell membranes, allowing H+ back-diffusion
- Toluene inhalation — occupational or recreational solvent exposure
Genetic dRTA: Autosomal dominant forms involve SLC4A1 mutations affecting AE1 trafficking. Autosomal recessive forms involve ATP6V1B1 (H+-ATPase B1 subunit, associated with sensorineural deafness) or ATP6V0A4 (H+-ATPase a4 subunit, usually without deafness). Genetic dRTA typically presents in infancy or early childhood with failure to thrive and rickets.
Type 2: Proximal RTA and Fanconi Syndrome
Type 2 (proximal) RTA results from impaired bicarbonate reabsorption in the proximal convoluted tubule (PCT). Under normal conditions, the PCT reclaims approximately 80–90% of filtered HCO3 via the sodium-hydrogen exchanger NHE3 (apical) and the sodium-bicarbonate cotransporter NBC1 (basolateral), both dependent on carbonic anhydrase II within the cell and carbonic anhydrase IV on the apical surface. Dysfunction of any of these transporters allows bicarbonate to flood the distal tubule in amounts that exceed its limited reabsorptive capacity, and bicarbonate is lost in urine.
This bicarbonate wasting continues until the serum HCO3 falls to a new, lower "threshold" level — typically 14–18 mEq/L — at which point the filtered HCO3 load is low enough for the proximal tubule to handle. Below this threshold, urine pH paradoxically normalizes (can fall below 5.5), a feature that distinguishes Type 2 from Type 1 RTA and can cause diagnostic confusion during acute presentations.
Volume depletion from bicarbonate and sodium loss activates the renin-angiotensin-aldosterone axis, driving potassium excretion in the collecting duct and producing hypokalemia — just as in Type 1, but through a different mechanism.
Fanconi syndrome describes global proximal tubule dysfunction — the simultaneous failure to reabsorb not only bicarbonate but also phosphate, glucose (glucosuria at normal blood glucose levels), amino acids (aminoaciduria), uric acid, and potassium. This "everything leaks" phenotype occurs when a toxic or infiltrative process damages the PCT more broadly than the isolated bicarbonate transporters. Major causes include:
- Multiple myeloma — free light chains (especially kappa) are directly toxic to PCT cells; the nephrotoxic form is called "myeloma kidney" or "cast nephropathy"
- Tenofovir (antiretroviral, TDF/TAF) — inhibits mitochondrial DNA polymerase gamma in PCT cells, impairing oxidative phosphorylation; leading iatrogenic cause; all HIV patients on tenofovir should have annual urine phosphate and glucose monitoring
- Wilson's disease — copper deposition in PCT cells
- Cystinosis — lysosomal cystine accumulation damages PCT cells; the classic pediatric cause
- Galactosemia and hereditary fructose intolerance — metabolite accumulation in PCT cells
- Heavy metal poisoning — lead, cadmium, mercury
- Acetazolamide — carbonic anhydrase inhibitor; iatrogenic Type 2 RTA
- Ifosfamide (chemotherapy)
Type 4: Hyperkalemic RTA
Type 4 RTA is the most common form of RTA in clinical practice. Unlike Types 1 and 2, it produces hyperkalemia rather than hypokalemia, and the acidosis is typically mild (serum HCO3 rarely below 17 mEq/L). The unifying defect is insufficient aldosterone activity — either from impaired production (hypoaldosteronism) or resistance — in the collecting duct principal cells. Aldosterone normally drives sodium reabsorption via ENaC channels, creating the lumen-negative electrical gradient that drives both potassium secretion via ROMK and hydrogen ion secretion via H+-ATPase. Without this gradient, both K+ and H+ accumulate in the blood.
Type 4a — Hypoaldosteronism:
- Hyporeninemic hypoaldosteronism (most common cause overall) — occurs in elderly patients with diabetic nephropathy or chronic interstitial nephritis; damaged juxtaglomerular apparatus produces insufficient renin → inadequate angiotensin II → inadequate aldosterone; serum renin low, aldosterone low; GFR is typically mildly to moderately reduced (CKD stage 3); the combination of low renin and low aldosterone distinguishes this from primary adrenal failure
- Addison's disease (primary adrenal insufficiency) — destruction of adrenal cortex; both glucocorticoid and mineralocorticoid deficiency; renin elevated, aldosterone undetectable; life-threatening if acute
- Isolated mineralocorticoid deficiency — rare; congenital (CYP11B2 mutations) or acquired
Type 4b — Aldosterone Resistance (Pseudohypoaldosteronism):
- Trimethoprim and pentamidine — block ENaC channels in the collecting duct, mimicking aldosterone deficiency; trimethoprim is a common cause of unexplained hyperkalemia in patients on high-dose trimethoprim-sulfamethoxazole for Pneumocystis pneumonia prophylaxis
- Amiloride and triamterene — potassium-sparing diuretics that directly block ENaC
- Spironolactone and eplerenone — aldosterone receptor antagonists; can cause Type 4 in patients with CKD where potassium excretion is already compromised
- ACE inhibitors and ARBs — reduce angiotensin II → reduce aldosterone; Type 4 risk is especially high in CKD and heart failure patients on RAAS blockade
- Heparin and low-molecular-weight heparin — directly suppress adrenal aldosterone synthesis
- Pseudohypoaldosteronism Type 1 (PHA1) — genetic ENaC mutations (autosomal recessive, severe) or mineralocorticoid receptor mutations (autosomal dominant, milder)
Diagnosis and Laboratory Findings
The first step is recognizing hyperchloremic non-anion-gap metabolic acidosis on basic metabolic panel: low serum HCO3 with normal (or elevated) serum chloride, anion gap within the normal range (8–12 mEq/L). This pattern immediately excludes lactic acidosis, diabetic ketoacidosis, and uremic acidosis, pointing toward RTA or GI bicarbonate loss (diarrhea).
Urine pH is the critical initial discriminator:
- Urine pH persistently >5.5 in the setting of systemic acidosis = strongly suggests Type 1 dRTA (collecting duct cannot acidify)
- Urine pH <5.5 with acidosis = compatible with Type 2 (below the bicarbonate threshold, the distal nephron acidifies normally) or Type 4
Serum potassium is the second major discriminator:
- Hypokalemia → Type 1 or Type 2 RTA
- Hyperkalemia → Type 4 RTA
Urine anion gap (UAG) estimates urinary ammonium excretion, the primary mechanism of net acid excretion: UAG = Urine Na + Urine K − Urine Cl.
- Positive UAG (low ammonium excretion) → impaired renal acid excretion → RTA (Types 1 or 4)
- Negative UAG (high ammonium) → normal renal acidification; acidosis is likely from GI bicarbonate loss (diarrhea)
Additional targeted studies:
- Urine glucose and amino acids (glucosuria at normal blood glucose + aminoaciduria = Fanconi syndrome → Type 2)
- Urine phosphate, uric acid (wasting in Fanconi)
- Serum and urine protein electrophoresis (myeloma screen for Type 2)
- Plasma renin activity and serum aldosterone (both low in hyporeninemic hypoaldosteronism; renin high + aldosterone low in Addison's)
- Renal ultrasound (medullary nephrocalcinosis in Type 1 — bilateral, echogenic renal medulla)
- Autoimmune panel: ANA, anti-SSA/SSB (Sjogren's), complement (SLE)
- Serum cortisol and ACTH stimulation test (if Addison's suspected)
Differential Diagnosis
Non-anion-gap metabolic acidosis has a finite differential that can be systematically worked through:
- Diarrhea: The most common cause of hyperchloremic acidosis in outpatients. GI bicarbonate loss is distinguished from RTA by a negative urine anion gap (high ammonium excretion, intact renal acidification) and the clinical history. Urine pH is appropriately acid (<5.5).
- Bartter syndrome and Gitelman syndrome: These renal tubular disorders cause hypokalemia and alkalosis (not acidosis), so they are not typically confused with RTA; however, the clinical context of renal tubular dysfunction and electrolyte disturbances overlaps when reviewing charts.
- Ureterosigmoidostomy / GI diversions: Sigmoid colon reabsorbs ammonium chloride from urine, generating hyperchloremic acidosis; urine is absent from bladder so urine studies are not helpful.
- Early CKD: Mild CKD (GFR 30–60) can produce mild non-anion-gap acidosis from reduced ammoniagenesis and impaired net acid excretion; as GFR falls further, unmeasured anions accumulate and the anion gap rises.
- Exogenous acid loads: Large volume normal saline infusion (hyperchloremic acidosis from chloride load), ammonium chloride ingestion, acetazolamide.
- Adrenal insufficiency (Addison's disease): Presents with hyperkalemia and metabolic acidosis identical to Type 4 RTA, but accompanied by hyponatremia, hypotension, hyperpigmentation, and elevated ACTH. Morning cortisol and ACTH stimulation test are diagnostic.
Treatment and Management
Type 1 Distal RTA:
- Potassium citrate (first choice): Corrects both the acidosis (citrate is metabolized to bicarbonate) and the hypokalemia (potassium salt) simultaneously; reduces hypercalciuria by raising urinary citrate, which chelates calcium and inhibits calcium phosphate crystal growth; prevents new stone formation and may dissolve existing calcium phosphate stones; typical dose 1–2 mEq/kg/day in 2–3 divided doses
- Sodium bicarbonate: Alternative for patients unable to tolerate citrate; does not correct hypokalemia; 1–3 mEq/kg/day
- Monitoring: Serial renal ultrasound (nephrocalcinosis), urine calcium-to-creatinine ratio, serum potassium and HCO3, GFR
- Treat secondary cause: Immunosuppression for Sjogren's/SLE-related dRTA
Type 2 Proximal RTA:
- Alkali supplementation: Requires much higher doses than Type 1 (often 10–15 mEq/kg/day in children) because bicarbonate wasting resumes with any rise in serum HCO3 above the lowered threshold; potassium citrate preferred for the hypokalemia component
- Thiazide diuretics: Paradoxically reduce bicarbonate wasting by causing mild volume depletion, which increases proximal tubule sodium (and thus bicarbonate) reabsorption; used adjunctively to reduce alkali dose requirements
- Phosphate supplementation: In Fanconi syndrome with symptomatic phosphaturia-related hypophosphatemia and osteomalacia
- Vitamin D supplementation: For phosphate-wasting rickets/osteomalacia
- Treat underlying cause: Stop offending drug (tenofovir → switch to abacavir-based regimen); treat myeloma; copper chelation for Wilson's disease
Type 4 Hyperkalemic RTA:
- Fludrocortisone (0.05–0.2 mg/day): Synthetic mineralocorticoid; effective for true aldosterone deficiency (Addison's disease, hyporeninemic hypoaldosteronism) — corrects both hyperkalemia and acidosis; monitor blood pressure and edema (may worsen hypertension/heart failure)
- Dietary potassium restriction: Avoid high-potassium foods; reinforced in patients with CKD where excretion is already limited
- Loop diuretics (furosemide, bumetanide): Increase distal sodium delivery, which stimulates aldosterone-independent potassium secretion; useful in volume-overloaded or hypertensive patients where fludrocortisone is contraindicated
- Sodium bicarbonate: Addresses the acidosis component; may also reduce hyperkalemia by driving potassium intracellularly
- Discontinue offending drugs: Trimethoprim, amiloride, ACE inhibitors/ARBs, heparin; re-evaluate risk-benefit in critical medications
- Potassium binders (patiromer, sodium zirconium cyclosilicate): For persistent hyperkalemia in patients who cannot stop RAAS blockade
Prognosis and Long-Term Outcomes
With appropriate alkali therapy, the prognosis of all three forms of RTA is generally favorable. Children with genetic Type 1 or Type 2 RTA who receive prompt treatment achieve normal growth and do not develop chronic kidney disease from the acidosis itself. Key long-term concerns vary by type:
- Type 1 — nephrocalcinosis and stone disease: If untreated, progressive medullary calcium phosphate deposition can reduce GFR over years to decades. Potassium citrate therapy effectively prevents new stone formation and stabilizes or reduces nephrocalcinosis in most patients. Established nephrocalcinosis rarely fully resolves. Periodic renal ultrasound every 1–2 years is standard.
- Type 1 — bone disease: Chronic acidosis mobilizes skeletal calcium carbonate as a buffer; long-term consequences include osteopenia, osteoporosis, and fracture risk, particularly in adults diagnosed late. Alkali correction largely prevents further bone loss.
- Type 2 — underlying disease dominates prognosis: In acquired Type 2/Fanconi syndrome, the prognosis is largely determined by the underlying cause (myeloma, cystinosis, HIV-associated tenofovir nephrotoxicity). Removal of the offending agent — especially tenofovir — is often followed by partial recovery of proximal tubular function.
- Type 4 — CKD progression: Hyporeninemic hypoaldosteronism occurs in the setting of diabetic nephropathy and CKD; the underlying kidney disease drives GFR trajectory. Correction of the hyperkalemia with loop diuretics or fludrocortisone allows continuation of RAAS blockade, which itself is renoprotective in diabetic nephropathy — creating a treatment synergy.
- Cardiac arrhythmia risk: Both severe hypokalemia (Type 1/2) and severe hyperkalemia (Type 4) can cause life-threatening ventricular arrhythmias; monitoring serum potassium is the single most critical safety parameter in ongoing RTA management.
Key Research Papers
- Search PubMed — Rodriguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002.
- Search PubMed — Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012.
- Search PubMed — Sharma S, Bhatt H. Distal renal tubular acidosis: a systematic approach. Pediatr Nephrol. 2016.
- Search PubMed — Igarashi T et al. Mutations of the carbonic anhydrase II gene in patients with proximal renal tubular acidosis. Kidney Int. 1992.
- Search PubMed — Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol. 2014.
- Search PubMed — Kamel KS, Halperin ML. Intrarenal urea recycling leads to a higher rate of renal excretion of potassium: a new hypothesis with clinical implications. Curr Opin Nephrol Hypertens. 2011.
- Search PubMed — Fuster DG, Alexander RT. Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflugers Arch. 2014.
- Search PubMed — Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant. 2012.
- Search PubMed — Laing CM, Toye AM, Capasso G, Unwin RJ. Renal tubular acidosis: developments in our understanding of the molecular basis. Int J Biochem Cell Biol. 2005.
- Search PubMed — Carvalho M et al. Kidney stone risk and nephrocalcinosis in children with distal renal tubular acidosis. Pediatr Nephrol. 2013.
- Search PubMed — Nicolaidou P et al. Clinical outcomes in patients with distal renal tubular acidosis and nephrocalcinosis. Nephron. 2018.
- Search PubMed — Weiner ID, Linas SL, Wingo CS. Disorders of potassium metabolism. J Intensive Care Med. 2014.
Connections
- Nephrology & Hepatology
- The Sodium–Potassium Pump — interactive animation
- Bartter Syndrome — renal tubular hypokalemia + alkalosis; key differential for dRTA
- Gitelman Syndrome — distal tubular salt-wasting with hypokalemia; compare to Type 1 RTA
- Nephrolithiasis — calcium phosphate stones are the hallmark complication of Type 1 RTA
- Chronic Kidney Disease — Type 4 RTA typically occurs in advanced diabetic CKD
- Addison's Disease — primary adrenal insufficiency is a major cause of Type 4 RTA
- Sjogren's Syndrome — most common secondary cause of Type 1 distal RTA
- Potassium — hypokalemia (Types 1 and 2) and hyperkalemia (Type 4) are central electrolyte disturbances
- Calcium — chronic acidosis mobilizes skeletal calcium, driving the hypercalciuria, nephrocalcinosis, and bone loss of Type 1 RTA