Gitelman Syndrome
Most common inherited renal tubular disorder (prevalence ~1:40,000); autosomal recessive; SLC12A3 mutation causing defective thiazide-sensitive NaCl cotransporter (NCC) in distal convoluted tubule (DCT).
Table of Contents
- Overview and Pathophysiology
- The Biochemical Tetrad
- Clinical Presentation
- Diagnosis and Work-Up
- Differentiating Gitelman from Bartter Syndrome
- Cardiovascular Risks and QT Prolongation
- Treatment and Management
- Prognosis and Quality of Life
- Key Research Papers
- Connections
Overview and Pathophysiology
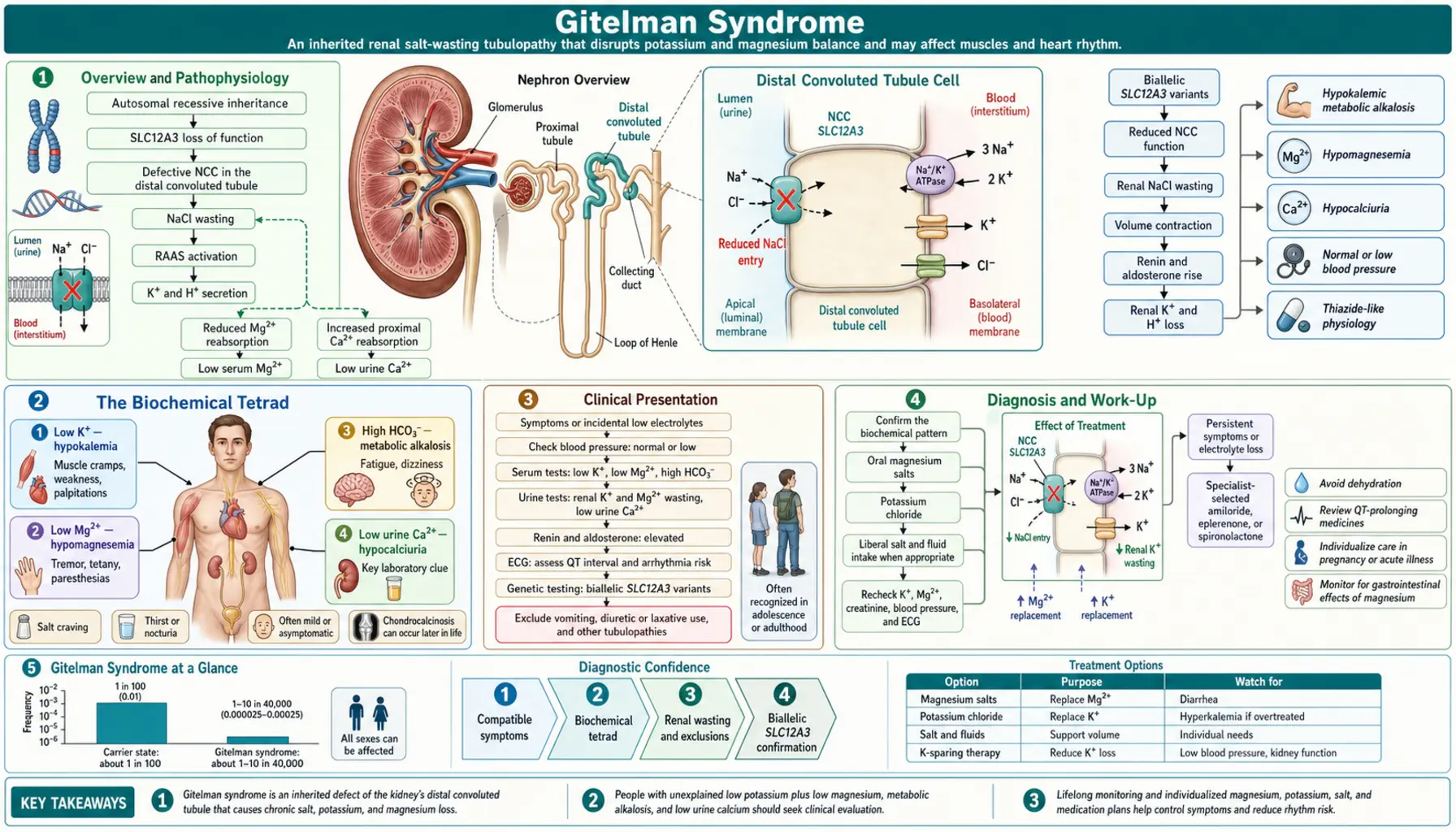
Gitelman syndrome is the most common inherited salt-losing tubulopathy, caused by loss-of-function mutations in SLC12A3, which encodes the thiazide-sensitive NaCl cotransporter (NCC) in the distal convoluted tubule (DCT). The DCT reabsorbs approximately 5–7% of filtered NaCl. NCC inactivation leads to a cascade of downstream electrolyte disturbances:
- Salt wasting → volume depletion → RAAS activation → secondary aldosteronism → increased ENaC activity → K+ and H+ excretion → hypokalemia + metabolic alkalosis
- Reduced luminal Na+ → increased Na/Ca exchanger activity on basolateral membrane → increased intracellular Ca2+ pumped out → net hypocalciuria
- Impaired Mg2+ reabsorption via TRPM6 (Mg2+ channel in DCT, NCC-dependent) → hypomagnesemia
The resulting phenotype closely mimics chronic thiazide diuretic use — this pharmacological parallel is central to understanding the mechanism and guides both diagnosis and treatment.
Inheritance is autosomal recessive. Heterozygous carriers are usually asymptomatic but may have mild electrolyte changes. More than 400 pathogenic variants in SLC12A3 have been identified; compound heterozygosity is common.
The Biochemical Tetrad
Gitelman syndrome is defined by four characteristic laboratory findings that together constitute the diagnostic biochemical profile:
- Hypokalemia: typically 2.5–3.2 mEq/L, with elevated urine potassium (inappropriate kaliuresis despite low serum K+, driven by aldosterone). The urinary K/Cr ratio is elevated, confirming renal potassium wasting rather than extrarenal losses.
- Metabolic alkalosis: elevated HCO3- (usually 28–36 mEq/L), arising from aldosterone-driven H+ secretion and from contraction alkalosis secondary to volume depletion.
- Hypomagnesemia: serum Mg2+ <0.7 mmol/L; caused by impaired TRPM6-mediated Mg2+ reabsorption in the DCT. Present in approximately 70% of patients. Clinically important because hypomagnesemia causes neuromuscular irritability and potentiates cardiac arrhythmias independent of potassium.
- Hypocalciuria: urine Ca/Cr ratio <0.1 mmol/mmol (or 24-hour urine calcium <2.5 mg/kg/day). This is the KEY DISTINGUISHING FEATURE from Bartter syndrome, which produces hypercalciuria. The mechanism: NCC inactivation increases basolateral Na/Ca exchange, enhancing transcellular Ca2+ reabsorption.
All four findings, combined with normal blood pressure, elevated plasma renin activity, and elevated aldosterone, constitute the complete diagnostic biochemical profile of Gitelman syndrome.
Clinical Presentation
Onset typically occurs in late childhood, adolescence, or adulthood — unlike Bartter syndrome, which often presents in infancy or early childhood. A substantial proportion of patients are diagnosed incidentally when routine laboratory panels reveal unexplained hypokalemia.
Symptomatic patients commonly report:
- Fatigue and general weakness — hypokalemia reduces skeletal muscle ATP production and impairs membrane repolarization
- Muscle cramps and tetany — hypomagnesemia increases neuromuscular irritability; alkalosis reduces ionized calcium, compounding this effect
- Salt craving — a behavioral response to chronic salt wasting; patients often describe an instinctive preference for salty foods
- Palpitations — hypokalemia prolongs cardiac action potential duration and QT interval; hypomagnesemia further destabilizes cardiac rhythm
- Paresthesias — tingling or numbness in extremities, attributable to hypomagnesemia and alkalosis-induced reduction in ionized calcium
- Nocturia and mild polyuria — present but less severe than in Bartter syndrome; reflects impaired urinary concentrating ability
- Normal blood pressure and normal growth — important distinguishing features; Bartter patients often exhibit failure to thrive and growth retardation
- Chondrocalcinosis — calcium pyrophosphate crystal deposition in cartilage, particularly in patients with long-standing hypomagnesemia; may cause acute pseudogout attacks
Gitelman syndrome does not cause polyhydramnios (excess amniotic fluid) or nephrocalcinosis, both of which are associated with antenatal Bartter syndrome.
Diagnosis and Work-Up
No single test diagnoses Gitelman syndrome — confirmation requires integration of clinical presentation, biochemical pattern, and genetic testing. A systematic approach:
- Step 1 — Confirm the biochemical tetrad: Serum K+, Mg2+, bicarbonate, and spot urine calcium-to-creatinine ratio (or 24-hour urine calcium). Hypocalciuria on a random urine specimen (Ca/Cr <0.1 mmol/mmol) is strongly suggestive.
- Step 2 — Confirm secondary hyperaldosteronism: Plasma renin activity (elevated) and serum aldosterone (elevated) in the absence of hypertension distinguishes Gitelman/Bartter from primary hyperaldosteronism (which suppresses renin).
- Step 3 — Exclude diuretic use: Urine thiazide and loop diuretic screen by mass spectrometry. Surreptitious diuretic use is the most important mimicker.
- Step 4 — Genetic testing: SLC12A3 sequencing identifies pathogenic variants in approximately 85–90% of clinically diagnosed cases. Next-generation sequencing panels covering SLC12A3 plus other tubulopathy genes are preferred. Some patients are compound heterozygotes with one allele carrying a deep intronic or splice-site variant.
- Step 5 — If SLC12A3 negative: Consider CLCNKB sequencing (Bartter Type III / mixed phenotype). Some CLCNKB mutations produce a Gitelman-like phenotype including hypocalciuria.
Historical note: The thiazide response test (thiazide causes less natriuresis in Gitelman patients than in healthy controls because the target NCC is already non-functional) was used prior to widespread genetic testing. It has been largely replaced by genetic analysis but remains a conceptual teaching tool.
Differentiating Gitelman from Bartter Syndrome
Gitelman and Bartter syndromes share hypokalemia, metabolic alkalosis, normal blood pressure, and elevated renin/aldosterone. The key distinguishing features are:
| Feature | Gitelman Syndrome | Bartter Syndrome (Types I/II/IV) | Bartter Type III |
|---|---|---|---|
| Urine calcium | Hypocalciuria (hallmark) | Hypercalciuria | Variable |
| Serum magnesium | Low (~70% of patients) | Usually normal | Usually normal |
| Age of onset | Adolescence/adulthood | Neonatal/early childhood | Childhood/adolescence |
| Clinical severity | Mild; often incidental | Severe; growth failure | Moderate |
| Nephrocalcinosis | Absent | Present (antenatal types) | Less common |
| Polyhydramnios | Absent | Present (Types I/II/IV) | Absent |
| Causative gene | SLC12A3 (DCT NCC) | SLC12A1, KCNJ1, BSND (TAL) | CLCNKB (TAL/DCT) |
Both conditions share elevated urine chloride (confirming renal Cl wasting rather than vomiting-related alkalosis, which causes low urine Cl). The hypocalciuria in Gitelman is arguably the single most reliable bedside biochemical discriminator.
Cardiovascular Risks and QT Prolongation
Although Gitelman syndrome is generally a mild condition, its electrolyte disturbances carry real cardiac risk that deserves attention in clinical management:
Hypokalemia and QT prolongation: K+ channels (particularly IKr, encoded by KCNH2) are responsible for phase 3 cardiac repolarization. When serum K+ falls, IKr current decreases, delaying repolarization and prolonging the QTc interval. A QTc >450 ms (males) or >460 ms (females) indicates elevated risk.
Hypomagnesemia: Mg2+ stabilizes cardiac ion channels. Hypomagnesemia independently prolongs QT and, more dangerously, predisposes to early afterdepolarizations — the trigger for torsades de pointes (TdP), a potentially fatal polymorphic ventricular tachycardia. The combination of hypoK + hypoMg is synergistically arrhythmogenic.
Drug interactions: Many commonly prescribed medications prolong QT (antipsychotics including haloperidol and quetiapine; macrolide antibiotics; fluoroquinolones; methadone; ondansetron). These drugs carry substantially increased arrhythmia risk in patients with uncorrected Gitelman electrolyte disturbances. Clinicians should screen for QT-prolonging drugs at every visit and prioritize electrolyte correction before prescribing them.
Clinical monitoring: ECG is recommended at baseline and during acute illness (when dehydration and vomiting can acutely worsen hypoK and hypoMg). Routine ECG monitoring at annual follow-up is reasonable in patients with persistent hypomagnesemia.
Treatment priority: Magnesium repletion should precede or accompany potassium repletion. Mg2+ is required for ROMK (renal outer medullary potassium) channel function — without adequate Mg2+, intracellular K+ continues to leak into the urine, making K+ supplementation less effective.
Treatment and Management
Treatment targets: serum K+ >3.5 mEq/L and serum Mg2+ >0.7 mmol/L. Complete normalization is often unachievable given ongoing renal wasting, but symptom control is the realistic goal.
1. Magnesium Supplementation (First Priority)
- Correct magnesium before or concurrently with potassium — Mg2+ is necessary for ROMK function; without it, supplemental K+ is rapidly excreted
- Preferred preparations: magnesium chloride or magnesium citrate (better absorbed than magnesium oxide); magnesium glycinate (best tolerated, least diarrhea)
- Typical dose: 300–600 mg elemental magnesium per day in divided doses (splitting doses reduces osmotic diarrhea)
- Approximately 20% of patients fail to achieve target Mg2+ despite supplementation due to ongoing renal wasting
2. Potassium Supplementation
- Oral potassium chloride (KCl) is preferred: 40–100 mEq/day, adjusted to response
- Sustained-release or microencapsulated formulations (e.g., K-Dur, Klor-Con) are better tolerated and reduce GI irritation
- IV potassium chloride for severe hypokalemia (<2.5 mEq/L), symptomatic episodes, or inability to tolerate oral therapy
3. Dietary Modifications
- High-sodium diet: counteracts ongoing salt wasting and reduces RAAS activation
- High-potassium foods: bananas, avocado, sweet potato, spinach, white beans
- Magnesium-rich foods: nuts (almonds, cashews), seeds (pumpkin, chia), legumes, dark chocolate, whole grains
4. Aldosterone Antagonists
- Spironolactone or eplerenone: block aldosterone at the collecting duct → reduce ENaC-mediated K+ and H+ secretion
- Useful adjunct when K+ supplementation alone is insufficient
- Monitor for hyperkalemia (uncommon at Gitelman baseline levels but possible with high doses)
5. Amiloride
- K+-sparing diuretic that directly blocks ENaC; also has modest Mg2+-retaining effect via TRPM6 upregulation
- Useful alternative or addition to aldosterone antagonists, particularly when Mg2+ repletion is the primary challenge
6. Indomethacin
- NSAIDs reduce prostaglandin-mediated renin secretion, thereby reducing aldosterone-driven losses
- Less commonly required in Gitelman than in Bartter syndrome; reserved for patients with persistent symptoms despite Mg/K supplementation
- Use with caution given renal and GI risks with long-term NSAID use
Prognosis and Quality of Life
The overall prognosis of Gitelman syndrome is generally favorable. Most patients lead full, productive lives with appropriate electrolyte supplementation.
Acute decompensation risk: GI illnesses involving vomiting or diarrhea can precipitate acute crises — additional fluid and electrolyte losses on top of the chronic baseline deficit can drive K+ below 2.0 mEq/L and Mg2+ below 0.4 mmol/L, with risk of muscle paralysis, tetany, and arrhythmia. Patients should have a written sick-day management plan and know when to seek emergency care.
Long-term renal function: Generally preserved. Gitelman syndrome does not cause nephrocalcinosis, and structural renal damage is not a characteristic feature. Renal function should be monitored periodically to detect unrelated comorbid renal disease.
Chondrocalcinosis: Chronic hypomagnesemia may contribute to calcium pyrophosphate (CPP) crystal deposition in cartilage, causing joint pain and episodic pseudogout. This can significantly impact quality of life and may require rheumatologic co-management.
Cardiac outcomes: Low risk with maintained electrolytes. Patients with persistent hypoK or hypoMg require closer monitoring and should avoid QT-prolonging drugs.
Pregnancy: Requires close electrolyte monitoring throughout gestation. Electrolyte disturbances may worsen due to volume expansion and increased renal demands. Neonatal outcomes are generally good; affected fetuses do not exhibit the polyhydramnios or prematurity seen in antenatal Bartter syndrome.
Recommended annual monitoring:
- Serum electrolytes (K+, Mg2+, HCO3-)
- 24-hour urine calcium and potassium
- Serum creatinine and eGFR
- ECG (especially if Mg2+ remains low or QT-prolonging drugs are prescribed)
- Blood pressure
Key Research Papers
- PMID 8696348 — Simon DB et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996.
- Search PubMed — Lemmink HH et al. Mutations in the gene encoding the thiazide-sensitive sodium chloride cotransporter in patients with Gitelman's syndrome. J Am Soc Nephrol. 1998.
- Search PubMed — Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008.
- Search PubMed — Blanchard A et al. Direction of net urine chloride flux as a biomarker of thiazide-sensitive sodium chloride cotransporter activity. Kidney Int. 2012.
- Search PubMed — Barakat AJ, Rennert OM. Gitelman syndrome. Am J Nephrol. 2014.
- Search PubMed — Walsh PR et al. Bartter and Gitelman syndromes. Orphanet J Rare Dis. 2018.
- Search PubMed — Hou J et al. Thiazide-sensitive NaCl cotransporter in Gitelman syndrome. Clin Kidney J. 2017.
- Search PubMed — Cruz DN et al. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001.
- Search PubMed — Vargas-Poussou R et al. Toward a comprehensive understanding of SLC12A3 mutations and Gitelman syndrome. J Am Soc Nephrol. 2017.
- Search PubMed — Blanchard A et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017.
- Search PubMed — Schlingmann KP et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004.
- Search PubMed — Riveira-Munoz E et al. Transcriptional and functional analyses of SLC12A3 mutations. J Am Soc Nephrol. 2007.
Connections
- Nephrology & Hepatology
- The Sodium–Potassium Pump — interactive animation
- Bartter Syndrome — similar salt-wasting tubulopathy affecting the thick ascending limb
- Nephrogenic Diabetes Insipidus — renal tubular dysfunction impairing water reabsorption
- Alport Syndrome — hereditary nephropathy affecting the glomerular basement membrane
- Magnesium — key supplementation target in Gitelman hypomagnesemia
- Potassium — hypokalemia management and repletion strategies
- Lab Tests — electrolyte panels, urine calcium, renin and aldosterone assays