Thalassemia: History and Discovery
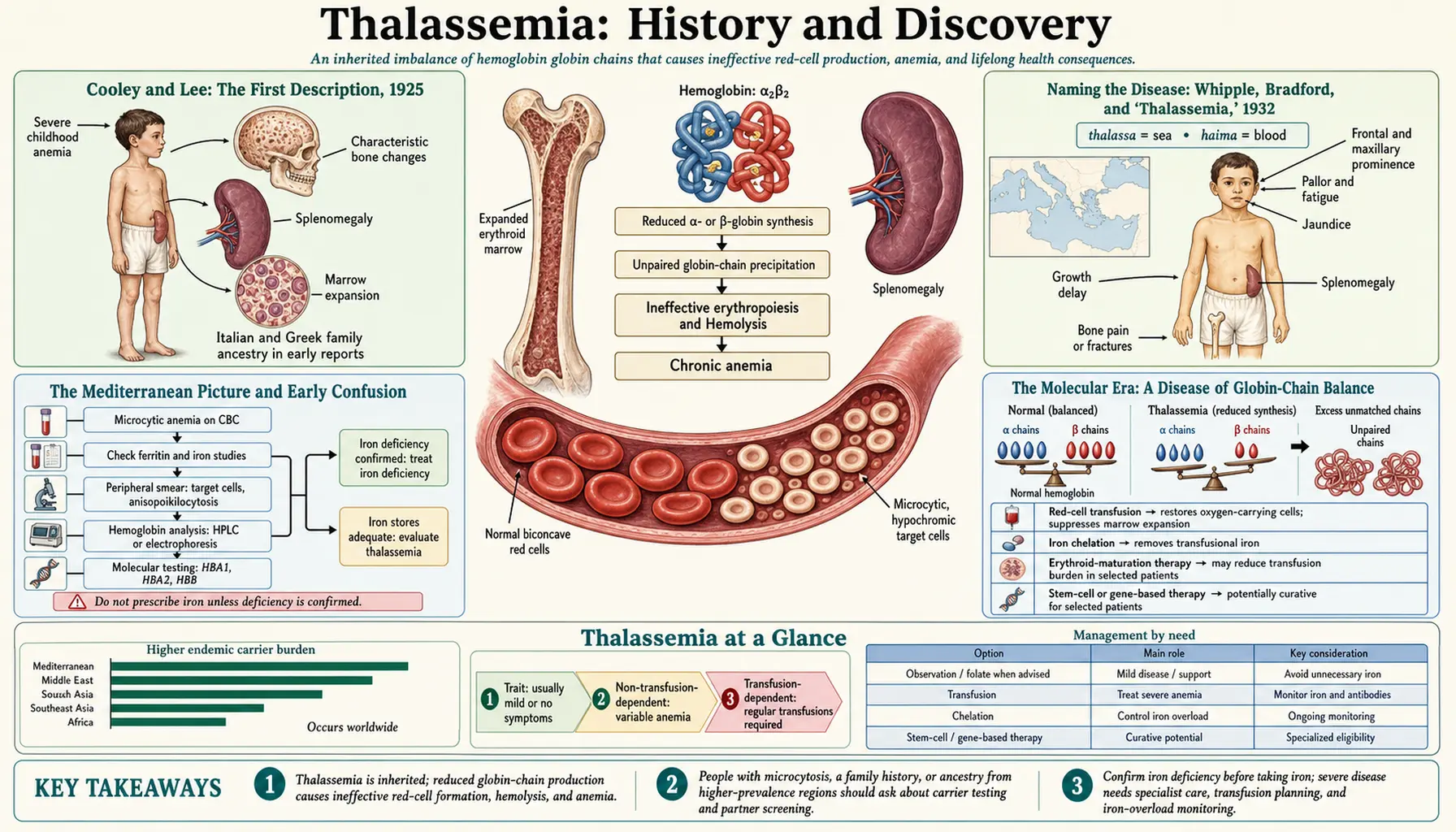
Thalassemia's story moves through three distinct discoveries that are easy to confuse and worth keeping separate. The first clinical description of the severe childhood form came in 1925, when Thomas Benton Cooley and Pearl Lee of Detroit reported a profound anemia with an enlarged spleen and striking bone changes, mostly in children of Italian, Greek, and other Mediterranean ancestry — the disorder still called Cooley's anemia. The name "thalassemia" — from the Greek thalassa (the sea, meaning the Mediterranean) and haima (blood) — was coined separately, in 1932, by George Hoyt Whipple and W. L. Bradford. The molecular cause — an imbalance between the alpha and beta globin chains of hemoglobin — was worked out only in the mid-twentieth century, making thalassemia one of medicine's first true "molecular diseases." This page traces that arc, distinguishes description from naming from mechanism, and is careful to mark hypothesis as hypothesis.
Table of Contents
- Cooley and Lee: The First Description, 1925
- Naming the Disease: Whipple, Bradford, and "Thalassemia," 1932
- The Mediterranean Picture and Early Confusion
- The Molecular Era: A Disease of Globin-Chain Balance
- Sorting Out Alpha and Beta Thalassemia
- The Malaria Hypothesis and Geographic Distribution
- Transfusion, Iron Chelation, and the Modern Era
- Gene Therapy and the Present Frontier
- Legacy: A Model Molecular Disease
- Research Papers and References
- Connections
- Featured Videos
Cooley and Lee: The First Description, 1925
The modern medical history of thalassemia begins in Detroit. Thomas Benton Cooley (1871–1945), a pediatrician associated with the Children's Hospital of Michigan, and his colleague Pearl Lee, presented a small series of children who shared a distinctive and severe picture: a profound anemia beginning in early childhood, a greatly enlarged spleen (splenomegaly), an enlarged liver, a sallow or muddy-yellow complexion, and unusual changes in the skull and facial bones. The report — commonly cited as "A series of cases of splenomegaly in children with anemia and peculiar bone changes" — was presented to the Transactions of the American Pediatric Society in 1925, and an expanded account (with Witwer and Lee) followed in the American Journal of Diseases of Children in 1927.
Cooley recognized that this was something new and hereditary, distinct from the nutritional anemias of the day. A telling clinical observation underpinned his thinking: the affected children were often well-nourished youngsters of Italian and Greek families living in Detroit, far from any region where malaria was endemic, so the severe anemia could not simply be blamed on infection or poor diet. The bone changes — an expansion of the marrow-filled spaces that gives the classic radiographic "hair-on-end" skull and the characteristic facial appearance — reflected the body's desperate, overdriven attempt to make red cells. Cooley initially called the condition erythroblastic anemia, a term emphasizing the immature red-cell precursors (erythroblasts) seen in the blood.
Because Cooley first delineated the severe, transfusion-requiring form of the disease — what is now understood as beta-thalassemia major — that form has carried his name ever since as Cooley's anemia. It is important to be precise about what 1925 represents: it is the first clear clinical description of the syndrome, not the coining of the word "thalassemia" (which came later) and not an understanding of its cause (which came much later still).
Naming the Disease: Whipple, Bradford, and "Thalassemia," 1932
The word thalassemia entered medicine in 1932, in a paper by George Hoyt Whipple and W. L. Bradford of the University of Rochester, published in the American Journal of Diseases of Children. Whipple — who would share the 1934 Nobel Prize in Physiology or Medicine for his work on anemia and liver therapy — and Bradford were dissatisfied with Cooley's label. As Whipple put it, they did not like the term "erythroblastic anemia," feeling that the immature red cells were not a sufficiently distinctive or defining feature of the condition to anchor its name.
What struck them instead was the disease's remarkable geography. In their words, the disease was "limited almost wholly to Italians, Greeks and Syrians" — that is, to peoples originating around the Mediterranean Sea. They therefore proposed a name built from that geographic fact: from the Greek thalassa (Θάλασσα), "the sea," long used specifically to denote the Mediterranean, combined with haima (αἷμα), "blood." The result, "thalassemia," can be read as "sea in the blood" or, more loosely, "the anemia of the Mediterranean people." Some historical accounts add that Whipple, classically educated, had in mind the famous cry of Xenophon's weary Greek soldiers on at last sighting the sea — "Thalassa! Thalassa!" While that anecdote is widely retold, the documented and load-bearing fact is the explicit Mediterranean rationale stated in the 1932 paper itself.
A point of historical nuance deserves an honest flag. Some secondary sources describe the term as having been first floated in the form "thalassic anemia" before settling on "thalassemia," and the precise spelling-and-credit history is occasionally debated; the primary sources consulted here document the 1932 Whipple-and-Bradford coinage and its Mediterranean rationale but do not independently confirm an original "thalassic anemia" spelling, so that detail is noted as a reported account rather than asserted as established fact. What is firmly established is the year (1932), the authors (Whipple and Bradford), the etymology (thalassa + haima), and the reasoning (the disease's confinement to Mediterranean populations).
The Mediterranean Picture and Early Confusion
For the first decades after Cooley's report, thalassemia was understood almost entirely as a clinical and ethnic phenomenon: a severe inherited anemia of Mediterranean children, sometimes called "Mediterranean anemia," "Cooley's anemia," or, after 1932, "thalassemia." Physicians could recognize the syndrome at the bedside and on radiographs long before anyone could say what was actually wrong inside the red cell. This is a recurring pattern in the history of medicine — the clinical entity is named and catalogued first, and the mechanism is filled in afterward.
Progress in the 1930s and 1940s sharpened the genetics. It became clear that thalassemia was inherited and that there was a spectrum of severity: a mild or essentially symptomless trait (thalassemia minor) carried by one parent, and a severe major form in children who inherited the abnormality from both parents. Researchers such as the Italian hematologists studying the disorder in its heartland, and others in Europe and the United States, established that the severe form behaved as if the child had received a "double dose" of the trait. This recognition of a carrier state and a homozygous severe state was a crucial bridge: it framed thalassemia as a quantitative, gene-dose disorder, which is exactly what the molecular era would confirm.
Still, through the first half of the twentieth century the fundamental question remained open. Why did these children fail to make enough normal hemoglobin? The answer would require a new science — the chemistry of the hemoglobin molecule itself — that did not yet exist when Cooley, Whipple, and Bradford were writing.
The Molecular Era: A Disease of Globin-Chain Balance
The decisive shift came in the 1950s and 1960s, when hemoglobin became one of the first human proteins understood in fine molecular detail. The pivotal conceptual breakthrough was the recognition that the adult hemoglobin molecule is built from two kinds of protein chains — two alpha globin chains and two beta globin chains — that must be produced in balanced, roughly equal amounts and then assembled into the functioning, oxygen-carrying tetramer. In 1956, Vernon Ingram famously pinned down the exact chemical difference between normal hemoglobin and sickle-cell hemoglobin (a single amino-acid substitution), launching the idea that inherited blood disorders could be precisely chemical — the concept of the "molecular disease."
Thalassemia turned out to be a different kind of molecular defect from sickle cell, and that contrast was clarifying. In sickle-cell disease the globin chain is made in normal amounts but is structurally abnormal. In thalassemia, by contrast, the chain's structure is usually normal, but it is made in reduced quantity or not at all — a problem of amount rather than shape. The core insight, developed through the globin-chain-synthesis studies of the 1960s, is that thalassemia is fundamentally a disease of imbalance: when too little beta chain is produced (beta-thalassemia), the now-excess alpha chains have no partner, precipitate inside developing red cells, and destroy those cells in the bone marrow before they ever mature — a process called ineffective erythropoiesis. The mirror situation holds for alpha-thalassemia. Investigators including David Weatherall, John Clegg, Hermann Lehmann, and others were central to demonstrating this chain-imbalance mechanism directly in patients' blood.
This was a profound reframing. The bone changes Cooley had drawn in 1925, the massive spleen, the lifelong transfusion dependence — all of it could now be traced back to a single quantitative failure: not enough of one globin chain, too much of the other, and red-cell precursors dying as a result. The clinical portrait of 1925 and the molecular explanation of the 1960s had finally met.
Sorting Out Alpha and Beta Thalassemia
Once thalassemia was understood as a globin-chain-balance disorder, it naturally split into two great families defined by which chain is deficient. Beta-thalassemia — the form Cooley first described — arises from reduced or absent production of the beta globin chain, and its severe homozygous form (beta-thalassemia major, Cooley's anemia) produces the classic transfusion-dependent childhood disease. Alpha-thalassemia arises from reduced production of the alpha globin chain and has its own distinctive forms, including the condition once cataloged simply as "hemoglobin H disease."
A key piece of evidence came from abnormal hemoglobins that appear when one chain is in short supply and the leftover chains pair up with themselves. When alpha chains are scarce, excess beta (and fetal gamma) chains assemble into unusual hemoglobins — hemoglobin H (four beta chains) and, in fetuses and newborns, hemoglobin Bart's (four gamma chains). The identification of these "orphan-chain" hemoglobins, including work by Hunt and Lehmann in 1959 characterizing hemoglobin Bart's as a fetal hemoglobin lacking alpha chains, gave investigators a direct chemical fingerprint of alpha-chain deficiency and helped cement the alpha-versus-beta framework.
This alpha/beta distinction is not academic bookkeeping. It governs how the disease presents, how severe it is, how it is inherited (the alpha globin genes are duplicated, so there are normally four alpha genes to lose, which is why alpha-thalassemia comes in graded severities), and how it is diagnosed. The reference text that synthesized this entire field, Weatherall and Clegg's The Thalassaemia Syndromes, became the standard work and ran through multiple editions over the following decades.
The Malaria Hypothesis and Geographic Distribution
One puzzle haunted thalassemia from the start: if the severe homozygous form is so devastating and so often fatal in childhood, why is the underlying gene so common in certain populations? Natural selection should, over time, weed out a gene that kills children before they reproduce. The leading explanation is the celebrated malaria hypothesis, proposed by the British geneticist J. B. S. Haldane in 1949. Haldane suggested that while inheriting two copies of a thalassemia gene is harmful, inheriting a single copy — being a carrier, with thalassemia trait — might confer partial protection against malaria, giving carriers a survival advantage in malarial regions. This is the same logic of "heterozygote advantage" that explains the geographic overlap of sickle-cell disease with historic malaria.
It is worth being clear about its status: Haldane's 1949 proposal was, and is properly described as, a hypothesis — a brilliant inference rather than, at the time, a proven fact. In the decades since, a large body of population-genetic and epidemiological evidence has accumulated supporting it, showing that both alpha- and beta-thalassemia traits can confer partial protection against severe Plasmodium falciparum malaria and that the global map of thalassemia closely tracks the historical map of malaria across the Mediterranean, the Middle East, South and Southeast Asia, and sub-Saharan Africa. The hypothesis is now strongly supported, but it began as exactly that, a hypothesis, and the strength of the modern evidence is itself part of the story.
The malaria hypothesis elegantly closes a circle that runs back to 1925. Cooley had noted that his Detroit patients were Mediterranean children living far from malaria — an observation that, decades later, points straight at why their ancestors' populations carried the gene in the first place. Geography named the disease (the "sea" of thalassemia) and geography, through malaria, also explains its distribution.
Transfusion, Iron Chelation, and the Modern Era
For most of the twentieth century, the mainstay of care for severe beta-thalassemia was regular blood transfusion. Transfusions correct the anemia and suppress the body's frantic, bone-deforming overproduction of red cells, transforming what had been a lethal childhood disease into a survivable chronic condition. But transfusion created a new and lethal problem of its own. Each unit of donated blood carries iron, and the human body has no efficient way to excrete a surplus. Over years of transfusions, iron accumulates relentlessly in the heart, liver, and endocrine glands — transfusional iron overload — and, untreated, this iron toxicity (especially to the heart) became the leading cause of death in well-transfused patients.
The answer was iron chelation: drugs that bind excess iron so it can be excreted. The first and long-dominant chelator, desferrioxamine (deferoxamine), was isolated from a soil bacterium and entered clinical use in the 1960s, gaining acceptance as standard therapy over the following decade or so. Its great drawback was practicality — it had to be given by slow, prolonged subcutaneous infusion, often overnight by pump, several nights a week, for life, which made adherence genuinely hard. The later development of effective oral iron chelators (deferiprone and deferasirox) was therefore a major quality-of-life advance, and combination regimens further improved control of iron loading. Together, transfusion plus chelation turned beta-thalassemia major from a disease of early childhood death into one in which many patients live well into adulthood.
For selected patients, allogeneic hematopoietic stem-cell (bone-marrow) transplantation from a matched donor offered something the transfusion-and-chelation regimen could not: an actual cure, by replacing the patient's blood-forming cells with healthy donor cells. The catch was the need for a suitable matched donor and the real risks of the transplant procedure itself, which limited how many patients could benefit.
Gene Therapy and the Present Frontier
The most recent chapter brings the story full circle, from describing a disease in 1925 to correcting its genetic root nearly a century later. Because thalassemia is caused by a defect in producing a specific globin chain, it has long been an obvious target for gene therapy — the idea of fixing or compensating for the faulty instructions in a patient's own blood-forming stem cells, then returning those cells to the patient.
Two distinct approaches reached approval in the 2020s. The first, betibeglogene autotemcel (marketed as Zynteglo), is a one-time therapy that adds a working, modified beta-globin gene into the patient's own harvested stem cells using a viral vector; the corrected cells are then reinfused so the patient can make functional hemoglobin. The U.S. Food and Drug Administration approved it in August 2022 for transfusion-dependent beta-thalassemia, making it the first gene therapy approved for the condition. The second approach is gene editing: exagamglogene autotemcel (Casgevy), which uses CRISPR-Cas9 to switch back on the body's production of protective fetal hemoglobin. First approved in late 2023 for sickle-cell disease, it received FDA approval for transfusion-dependent beta-thalassemia in January 2024 — among the first CRISPR-based therapies ever authorized.
These therapies are extraordinary scientific achievements that can free some patients from lifelong transfusions, though they remain complex, costly, and not yet available or appropriate for everyone. They represent the current frontier rather than the end of the road, and research into broader, more accessible options continues. For day-to-day patient information on living with the condition, see the main Thalassemia page.
Legacy: A Model Molecular Disease
Few inherited disorders have taught medicine as much as thalassemia. Because mature red blood cells are easy to obtain and make essentially one product — hemoglobin — in enormous quantity, the inherited disorders of hemoglobin became among the very first human diseases that could be dissected at the molecular and then the genetic level. Lessons first learned in thalassemia and sickle cell — how a single defect changes a protein, how gene dosage shapes severity, how mutations disrupt the regulation of protein production — became foundational principles of human molecular genetics far beyond hematology.
The history also carries a quieter lesson about how medical knowledge is built. The three milestones are genuinely separate achievements by different people in different eras: Cooley and Lee described the disease at the bedside in 1925; Whipple and Bradford named it in 1932; and a generation of molecular hematologists — Ingram, Lehmann, Weatherall, Clegg, and many others — explained why it happens, while Haldane hypothesized in 1949 why the gene endured. Keeping these distinct — description, naming, mechanism, and evolutionary hypothesis — is not pedantry; it is how the history actually unfolded, and it guards against the common error of collapsing a century of work into a single tidy "discovery."
From a deforming, fatal childhood anemia of unknown cause to a precisely understood molecular disease now being addressed with gene editing, thalassemia stands as one of the clearest illustrations in all of medicine of how careful clinical observation, basic chemistry, population genetics, and modern biotechnology can, across a hundred years, converge on a single small molecule and the people who carry its altered form.
Research Papers and References
The references below combine the historical milestone papers with curated PubMed topic-search links into the thalassemia literature. The earliest milestone publications — Cooley and Lee's 1925 Transactions report and the 1932 Whipple and Bradford naming paper in the American Journal of Diseases of Children — predate digital indexing and are named here as historical primary sources; where a stable DOI or PMID was confidently verified it is linked directly, otherwise a PubMed topic search is provided. Each external link opens in a new tab.
- Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Transactions of the American Pediatric Society. 1925;37:29–30. (Historical primary source — first clinical description of the severe childhood anemia later called Cooley's anemia.)
- Whipple GH, Bradford WL. Mediterranean disease — thalassemia (erythroblastic anemia of Cooley). American Journal of Diseases of Children. 1932;44(2):336–365. (Historical primary source — coinage of the term "thalassemia.")
- Cooley TB, Witwer ER, Lee P. Anemia in children with splenomegaly and peculiar changes in the bones. American Journal of Diseases of Children. 1927;34:347–363. (Historical primary source — expanded clinical account.)
- Weatherall DJ. Thalassaemia: the long road from bedside to genome. Nature Reviews Genetics. 2004;5(8):625–631. — doi:10.1038/nrg1406
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. The Lancet. 2012;379(9813):373–383. — doi:10.1016/S0140-6736(11)60283-3
- Thomas Benton Cooley, Pearl Lee, and the history of Cooley's anemia / thalassemia — PubMed: history of Cooley's anemia and thalassemia
- Origin and naming of "thalassemia" (Whipple and Bradford, Mediterranean disease) — PubMed: thalassemia naming and Mediterranean disease
- Ingram VM, Stretton AOW. Genetic basis of the thalassaemia diseases. Nature. 1959;184:1903–1909. — PubMed: Ingram & Stretton, genetic basis of thalassaemia
- Globin-chain synthesis imbalance as the molecular basis of thalassemia (Weatherall, Clegg, and colleagues) — PubMed: globin-chain synthesis imbalance in thalassemia
- Hunt JA, Lehmann H. Haemoglobin "Bart's": a foetal haemoglobin without alpha chains. Nature. 1959;184:872–873. — PubMed: Hunt & Lehmann, haemoglobin Bart's
- Haldane JBS and the malaria hypothesis — heterozygote advantage and thalassaemia distribution — PubMed: Haldane malaria hypothesis and thalassaemia
- Iron chelation therapy and the treatment of thalassemia (deferoxamine, deferiprone, deferasirox) — PubMed: iron-chelation therapy in thalassemia
- Gene therapy for transfusion-dependent beta-thalassemia — betibeglogene autotemcel and CRISPR exagamglogene autotemcel — PubMed: gene therapy for beta-thalassemia
- The inherited disorders of hemoglobin as the first "molecular diseases" — PubMed: hemoglobinopathies as molecular diseases
External Authoritative Resources
- GeneReviews (NCBI Bookshelf) — Beta-Thalassemia and Alpha-Thalassemia
- MedlinePlus Genetics — Thalassemia (National Library of Medicine)
- PubMed — All research on the history of thalassemia
Connections
- Hematology
- Thalassemia (main page)
- All Conditions
- Sickle Cell Disease
- Anemia
- Hemochromatosis (Iron Overload)
- Hemophilia
- Polycythemia Vera