Hemochromatosis: History and Discovery
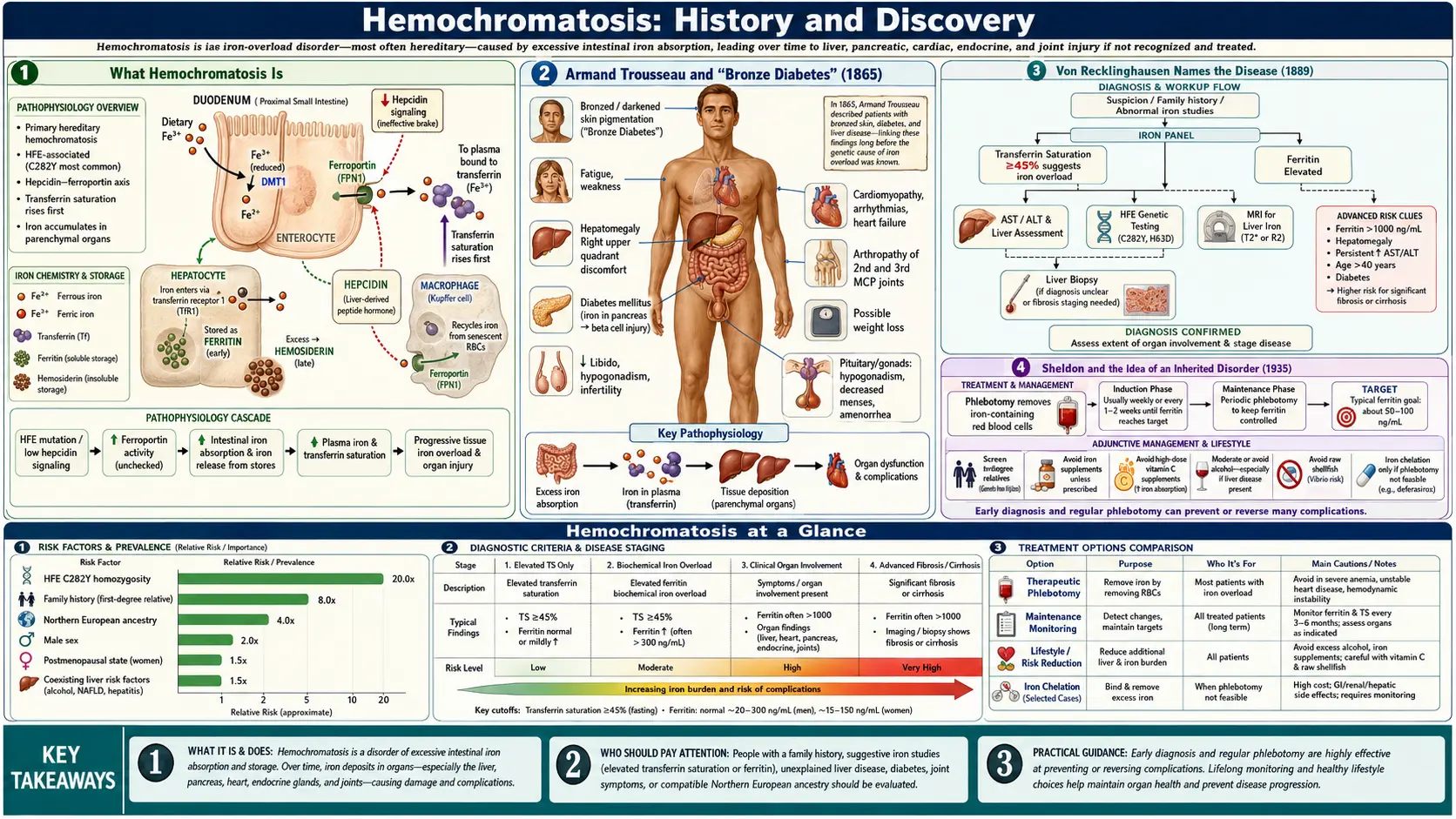
Hemochromatosis — the slow, silent overloading of the body with iron — was pieced together over more than a century by clinicians and pathologists who each saw only a part of the picture. The French physician Armand Trousseau described the haunting clinical triad of diabetes, bronze-grey skin, and a hardened liver in 1865, but he had no idea iron was to blame. The German pathologist Friedrich Daniel von Recklinghausen coined the very word “hemochromatosis” in 1889 after proving the liver pigment was iron — yet he guessed wrongly that the metal came from the blood. Only in 1935 did J. H. Sheldon argue the disease was inherited, and not until 1996 did James Feder and colleagues find the responsible HFE gene and its C282Y mutation, today recognized as one of the most common inherited conditions in people of Northern European descent. This is the story of how a “bronze diabetes” became a treatable genetic disease cured by the oldest remedy in medicine: removing blood.
Table of Contents
- What Hemochromatosis Is
- Armand Trousseau and “Bronze Diabetes” (1865)
- Von Recklinghausen Names the Disease (1889)
- Sheldon and the Idea of an Inherited Disorder (1935)
- The Rise of Therapeutic Phlebotomy
- Tracing It to the Genes: HLA and Chromosome 6
- The HFE Gene and the C282Y Mutation (1996)
- A Celtic Inheritance: The Most Common Genetic Disorder
- Legacy and Modern Understanding
- Research Papers and References
- Connections
- Featured Videos
What Hemochromatosis Is
Hemochromatosis is a disorder in which the body absorbs and stores too much iron. The human body has no active mechanism for excreting surplus iron, so once absorption runs unchecked, the metal accumulates year after year in the liver, pancreas, heart, joints, skin, and endocrine glands. Over decades this iron overload can produce cirrhosis of the liver, diabetes, heart failure, arthritis, a characteristic bronze or slate-grey skin tone, fatigue, and loss of libido. Because the build-up is slow and the early symptoms are vague, the disease historically declared itself only late, when organ damage was already advanced — which is exactly why its full nature took so long to understand.
The most common form, hereditary (or genetic) hemochromatosis, is an inherited condition in which a faulty gene causes the intestine to absorb iron as though the body were perpetually iron-deficient. Other forms arise secondarily — for example from repeated blood transfusions in disorders such as thalassemia, or from certain anemias — but it was the hereditary form, with its dramatic and consistent triad of signs, that first drew physicians’ attention. The history below traces how that triad was observed, how the iron was identified, how the inheritance was proven, and how the disease finally became a gene with a name.
It is worth keeping one thread in view throughout the story: at almost every stage, the people who advanced the science were working with incomplete or partly mistaken ideas. The disease was named for a belief about blood that turned out to be wrong; its standard treatment was adopted for a reason that was also wrong; and its true genetic cause remained hidden for more than a century after the first clinical description. Few diseases better illustrate how medicine moves forward by correcting itself.
Armand Trousseau and “Bronze Diabetes” (1865)
The clinical story begins in Paris in 1865 with Armand Trousseau (1801–1867), one of the most celebrated clinicians of nineteenth-century France and a gifted teacher whose Clinique médicale de l’Hôtel-Dieu de Paris shaped a generation of physicians. In a clinical lecture on diabetes, Trousseau presented the case of a man with the combination that would become the disease’s signature: diabetes, a hardened and pigmented (cirrhotic) liver, and a distinctive bronze-grey discoloration of the skin. This is generally regarded as the first description of what we now call hemochromatosis. (Trousseau’s name is also attached to several other discoveries, including Trousseau’s sign of latent tetany and the link between migratory thrombophlebitis and hidden cancer, known as Trousseau’s syndrome.)
Crucially, Trousseau did not know the cause. He did not associate the skin pigmentation or the liver scarring with iron — the chemistry to prove that did not yet exist in routine pathology, and the connecting idea simply had not been formed. He observed and recorded a striking pattern of disease; identifying its mechanism would fall to others. For accessibility, Trousseau’s lectures are named here as a historical primary source rather than cited as a modern paper.
In the years immediately following, the syndrome acquired its memorable popular labels. In 1871 the French pathologist Charles-Émile Troisier reported a case under the description diabète bronzé et cirrhose pigmentaire (“bronze diabetes and pigmentary cirrhosis”), crystallizing the triad in a single phrase. The vivid term “bronze diabetes” itself is widely credited to the French physician Victor Hanot, who used it in the 1880s; before the iron was understood, the condition was often simply called pigment cirrhosis. Whatever the exact attribution of the catchphrase, the clinical entity — bronze skin, sugar in the urine, and a scarred liver — was now firmly on the medical map, even though no one yet knew what the bronze pigment actually was.
Von Recklinghausen Names the Disease (1889)
The decisive next step was taken by the German pathologist Friedrich Daniel von Recklinghausen (1833–1910), a towering figure in nineteenth-century pathology (his name is also attached to von Recklinghausen disease, or neurofibromatosis type 1). In 1889 he examined the livers of patients who had died of “bronze diabetes” and, applying the iron-detecting staining methods developed by Max Perls and the framework of Rudolf Virchow’s cellular pathology, demonstrated that the mysterious brown pigment saturating the organs was in fact iron. This was the conceptual breakthrough: the bronze color was not melanin or blood breakdown in the ordinary sense, but massive deposition of iron throughout the tissues.
It was von Recklinghausen who coined the term “hämochromatose” (hemochromatosis) for the condition. The name encodes his theory of the disease: haemo- for blood and -chromatosis for a coloring or pigmentation, because he believed the iron-rich pigment was derived from the blood — that something circulating in or released from the blood was staining and damaging the organs. This is a notable and instructive error. We now know the iron in hereditary hemochromatosis comes from excessive absorption of dietary iron through the intestine, not from the breakdown of blood. The disease therefore carries, in its very name, a hypothesis that later proved wrong — yet the word has endured for well over a century.
A point sometimes blurred in popular accounts deserves clarity. Von Recklinghausen established that the pigment was iron and gave the disease its lasting name in 1889; in subsequent work around 1890 he and others advanced the understanding that iron infiltration of the pancreas could explain the associated diabetes. Even so, the full picture — that the iron overload was an inherited defect of iron absorption — would not emerge for decades. At the close of the nineteenth century the disease had a name and a culprit metal, but its origin remained a genuine mystery.
Sheldon and the Idea of an Inherited Disorder (1935)
For the first seventy years after Trousseau, hemochromatosis was regarded mainly as an acquired curiosity — variously blamed on alcohol, on the liver itself, or on some unknown toxin. The turning point in understanding came in 1935, when the British physician Joseph H. Sheldon published his landmark monograph simply titled Haemochromatosis (Oxford University Press). Sheldon undertook a comprehensive review of the medical literature, analyzing more than 300 reported cases (his series of 311 patients was overwhelmingly male), and from this synthesis he drew a conclusion that was ahead of its evidence.
Sheldon argued that hemochromatosis was best understood as an inborn error of metabolism — an inherited constitutional disorder of iron handling — rather than a consequence of alcohol or another external insult. In an era before molecular genetics, before the structure of DNA was known, this was a remarkably prescient inference, made purely from careful clinical reasoning and the familial clustering hinted at in the case reports. He effectively predicted, decades early, that the disease would prove to be genetic. Sheldon’s monograph is named here as a historical work; his central claim — that hemochromatosis is inherited — would be vindicated in full only at the end of the twentieth century.
Sheldon’s framing reoriented the field. If the disorder was a constitutional fault in metabolism, then the iron overload was the cause of the organ damage rather than an incidental finding, and the disease might in principle be present long before its late, devastating complications appeared. That logic pointed toward two of the most important developments still to come: a rational treatment aimed at removing iron, and, eventually, a search for the responsible gene.
The Rise of Therapeutic Phlebotomy
The treatment that transformed hemochromatosis from a fatal disease into a manageable one is also the oldest therapy in Western medicine: bloodletting. Removing blood removes red cells, and red cells are packed with iron in the form of hemoglobin; each unit of blood withdrawn carries off roughly 200–250 milligrams of iron. By bleeding a patient regularly over months and years, physicians could gradually deplete the enormous iron stores that the disease had accumulated — a treatment now called therapeutic phlebotomy or venesection.
There is a deep historical irony here that mirrors the naming of the disease. Phlebotomy was first applied to hemochromatosis partly on the mistaken premise that, since the iron was thought to come from the blood (as von Recklinghausen’s theory implied), removing blood ought to help. The premise was wrong — the iron comes from the gut, not from circulating blood — but the treatment works beautifully anyway, for a different reason: draining blood forces the body to draw on its stored iron to make new red cells, steadily unloading the overloaded organs. A wrong idea led to a right cure.
The value of repeated bleeding was put on a firm footing in 1950, when Davis and Arrowsmith reported “The effect of repeated bleeding in haemochromatosis” in the Journal of Laboratory and Clinical Medicine, documenting that systematic venesection could remove the excess iron and improve patients. Therapeutic phlebotomy remains the cornerstone of treatment to this day, and when it is started before irreversible organ damage develops, it can normalize life expectancy — making hemochromatosis one of the most satisfying chronic diseases to treat. (In a fitting modern twist, the blood removed from hemochromatosis patients is, where regulations permit, suitable for donation.)
Tracing It to the Genes: HLA and Chromosome 6
Sheldon had argued in 1935 that hemochromatosis was inherited, but proving it — and locating the responsible gene — required tools that did not yet exist. The breakthrough came in the mid-1970s from the French hematologist and geneticist Marcel Simon and his colleagues in Rennes. By studying families with the disease and typing their human leukocyte antigens (HLA) — the immune-system markers used in tissue matching — Simon’s group showed that hemochromatosis was strongly associated with particular HLA types (notably HLA-A3) and that it was inherited as an autosomal recessive trait, meaning a person needed two faulty copies of the gene, one from each parent, to develop the disease.
This HLA linkage was a pivotal clue for two reasons. First, it confirmed Sheldon’s inherited-disorder hypothesis with hard genetic data. Second, because the HLA genes are located on the short arm of chromosome 6, the discovery placed the unknown hemochromatosis gene in a specific neighborhood of the human genome. For the first time, researchers knew roughly where to look. Through the 1980s and early 1990s, family and population studies steadily refined the location, narrowing the search to a region near the HLA complex on chromosome 6p.
This period also brought practical diagnostic advances that no longer depended on finding the gene itself. Blood tests for serum iron and transferrin saturation, and later serum ferritin (a marker of total body iron stores), came into use through the 1950s, 1960s, and 1970s, allowing physicians to detect iron overload — and to screen relatives of patients — long before symptoms appeared. The stage was set for the final act: identifying the actual gene.
The HFE Gene and the C282Y Mutation (1996)
The hereditary basis of the disease was finally pinned down in 1996, when John (James) N. Feder and colleagues at the biotechnology company Mercator Genetics published their landmark paper, “A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis,” in Nature Genetics. Using positional-cloning methods to search the chromosome 6p region that the HLA studies had implicated, they identified a previously unknown gene — first called HLA-H and soon renamed HFE — lying near the HLA complex at chromosome 6p21.3. The gene encodes a protein resembling the MHC class I molecules of the immune system, which is now understood to help regulate how much iron the intestine absorbs.
The decisive finding was a single recurrent mutation. The great majority of the hemochromatosis patients Feder’s team studied carried, on both copies of HFE, a mutation now famous as C282Y — a change that substitutes the amino acid tyrosine for cysteine at position 282 of the HFE protein (the result of a single DNA base change at nucleotide 845). Loss of that cysteine disrupts a critical disulfide bond and cripples the protein’s normal function, releasing the brake on intestinal iron absorption. A second, milder mutation, H63D (histidine to aspartate at position 63), was identified in the same study and contributes to disease in some people, especially when paired with C282Y. In the original report, the large majority of typical patients were homozygous for C282Y — carrying two faulty copies — exactly fitting the autosomal recessive inheritance Simon had demonstrated two decades earlier.
The 1996 discovery was transformative. It confirmed Sheldon’s 1935 prediction, explained Simon’s HLA association, and — most importantly for patients — made it possible to diagnose hereditary hemochromatosis with a simple, definitive genetic blood test for the C282Y and H63D mutations. Relatives of an affected person could now be tested directly, and at-risk individuals could begin iron-removing phlebotomy before organ damage occurred. The original full citation is Feder JN, Gnirke A, Thomas W, et al., Nature Genetics, 1996;13(4):399–408.
A Celtic Inheritance: The Most Common Genetic Disorder
One of the most striking features of hereditary hemochromatosis is how common the C282Y mutation is in certain populations. It is most frequent in people of Northern European — particularly Irish, Scottish, Scandinavian, and other “Celtic” — ancestry, where carriers (people with one copy) are common and individuals with two copies (C282Y homozygotes, the group at risk of iron overload) occur at a frequency on the order of roughly 1 in 200 to 1 in 400. On this basis, HFE-related hereditary hemochromatosis is frequently described as the most common inherited (autosomal recessive) disorder among people of Northern European descent, and one of the most common genetic conditions in white populations of the United States.
The mutation’s concentration in north-western Europe has led to it being nicknamed the “Celtic mutation,” and researchers have proposed that the C282Y variant arose in a single ancestral population in that region thousands of years ago and spread with its descendants — a so-called founder effect. Several hypotheses have been offered for why such a potentially harmful gene became so widespread, including the idea that mildly enhanced iron absorption could have been an advantage in ancestral environments where dietary iron was scarce or iron-poor diets and blood loss were common. These remain reasonable but unproven hypotheses about the gene’s evolutionary history, and they should be read as informed speculation rather than established fact.
An equally important modern lesson is that of penetrance: many people who carry two copies of C282Y never develop serious iron overload or organ damage at all. The genetic mutation sets the stage, but sex (men are affected far more, and earlier, because menstruation protects many women through their reproductive years), diet, alcohol, blood loss, and other genetic and environmental factors all influence whether and how severely the disease appears. The gene is common; the full-blown disease is considerably less so — a nuance that distinguishes carrying the mutation from being ill with it.
Legacy and Modern Understanding
The history of hemochromatosis is a near-perfect parable of how medical knowledge accumulates. A great clinician (Trousseau, 1865) saw the disease without understanding it. A great pathologist (von Recklinghausen, 1889) identified the offending substance and named the disease — while attaching to it a theory about blood that was wrong. A thoughtful physician (Sheldon, 1935) reasoned his way to the idea of inheritance long before it could be proven. Geneticists (Simon and colleagues, 1970s) located the gene’s neighborhood, and finally molecular biologists (Feder and colleagues, 1996) found the gene itself and its C282Y mutation. Each generation corrected and completed the work of the last.
Modern understanding has continued well past 1996. In 2000–2001 the discovery of the iron-regulating hormone hepcidin revealed the master switch that governs how much iron the body absorbs and releases, and showed that HFE and related genes act, in effect, by disturbing hepcidin signaling — finally explaining at the molecular level why the intestine over-absorbs iron in this disease. Large population studies, including national biobank analyses, have since clarified how often C282Y homozygotes actually develop iron-related illness, sharpening the all-important distinction between carrying the gene and having the disease.
What makes this history genuinely hopeful is the destination. Hemochromatosis is now readily diagnosed — by iron-status blood tests and a definitive genetic test — and readily treated by the ancient remedy of removing blood, which, caught early, can give patients a normal life expectancy. A disease that once meant a slow death by bronze diabetes is today, in most cases, a manageable inherited condition. The long arc from a Paris lecture hall in 1865 to a genetic test on a tube of blood is a reminder that even the oldest mysteries in medicine can be solved — and that solving them changes lives.
Research Papers and References
The references below combine key peer-reviewed historical reviews and the landmark gene-discovery paper with curated PubMed topic-search links into the historical and genetic literature on hemochromatosis. The nineteenth-century primary sources (Trousseau’s clinical lectures, Troisier’s and Hanot’s case descriptions, and von Recklinghausen’s naming of the disease) are named in the article as historical sources rather than cited as modern papers. Each external link opens in a new tab.
- Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics. 1996;13(4):399–408. — doi:10.1038/ng0896-399
- Barton JC, Edwards CQ, Acton RT. A short history of hemochromatosis (and the broader history of iron overload). — PubMed: history of hemochromatosis (Trousseau, von Recklinghausen)
- Åsvold BO, et al. Hereditary haemochromatosis through 150 years. Tidsskrift for Den norske legeforening. 2016. — Tidsskrift: hereditary haemochromatosis through 150 years
- Hemochromatosis: Ancient to the Future (historical review). PMC, U.S. National Library of Medicine. — PMC7539181: Hemochromatosis — Ancient to the Future
- Sheldon JH. Haemochromatosis. Oxford University Press; 1935 (historical monograph proposing an inborn error of metabolism). — PubMed: Sheldon haemochromatosis (inborn error)
- Simon M, Bourel M, Genetet B, Fauchet R. HLA associations and the inheritance of idiopathic haemochromatosis (HLA-A3; chromosome 6). — PubMed: Simon, haemochromatosis and HLA linkage
- Davis WD, Arrowsmith WR. The effect of repeated bleeding in haemochromatosis. Journal of Laboratory and Clinical Medicine. 1950. — PubMed: Davis & Arrowsmith, repeated bleeding in hemochromatosis
- A history of phlebotomy (venesection) therapy for hemochromatosis. — PubMed: history of phlebotomy therapy for hemochromatosis
- Pietrangelo A. Hereditary hemochromatosis — pathogenesis, diagnosis, and treatment (HFE and iron biology). — PubMed: Pietrangelo, hereditary hemochromatosis (HFE)
- Origin and spread of the HFE C282Y mutation (the “Celtic” founder mutation in Northern Europeans). — PubMed: HFE C282Y Celtic founder mutation origin
- Penetrance of C282Y homozygosity (why many homozygotes never develop iron overload). — PubMed: C282Y homozygous HFE penetrance
- Discovery of hepcidin and the regulation of iron homeostasis (the molecular mechanism behind HFE disease). — PubMed: hepcidin and iron homeostasis in hemochromatosis
- Adams PC, et al. Population screening for HFE hemochromatosis (the HEIRS study and epidemiology). — PubMed: HEIRS study, HFE population screening
- General PubMed literature on the history and genetics of hereditary hemochromatosis. — PubMed: hereditary hemochromatosis history and HFE C282Y
External Authoritative Resources
- MedlinePlus Genetics — Hereditary Hemochromatosis
- GeneReviews (NCBI Bookshelf) — HFE Hemochromatosis
- PubMed — All research on hereditary hemochromatosis
Connections
- Hematology
- Hemochromatosis (Overview)
- All Conditions
- Iron (Essential Mineral)
- Anemia
- Thalassemia
- Cirrhosis
- Liver Disease
- Diabetes