Osteogenesis Imperfecta (Brittle Bone Disease)
- Overview and Classification
- Genetics: COL1A1, COL1A2, and Beyond
- Pathophysiology of Collagen Defects
- Clinical Features by Type
- Extra-Skeletal Manifestations
- Diagnosis
- Treatment and Management
- Emerging Therapies and Research
- Key Research Papers
- Connections
Overview and Classification
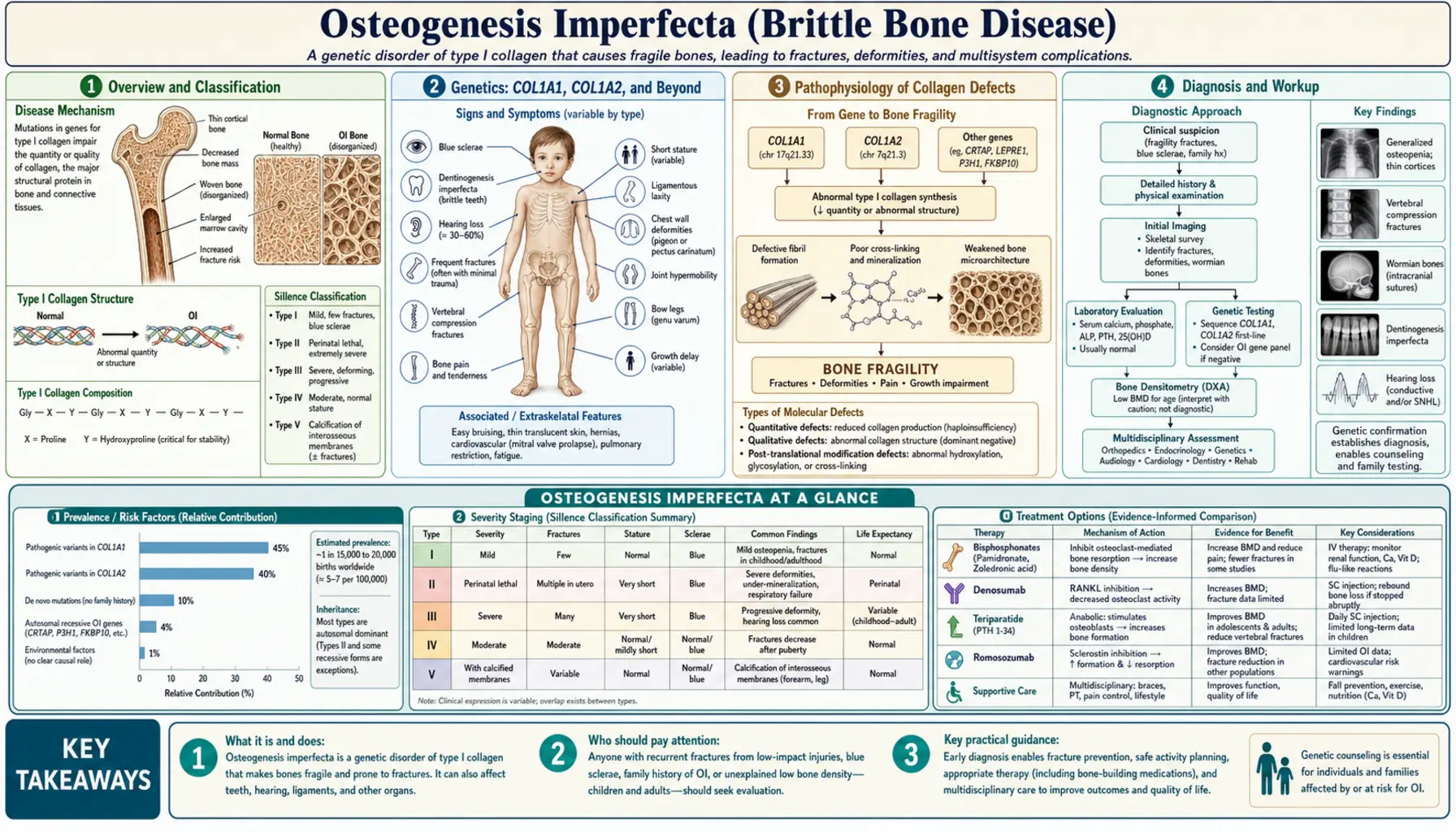
Osteogenesis imperfecta (OI), widely known as brittle bone disease, is a heritable connective tissue disorder caused primarily by defects in type I collagen — the protein that gives bone its tensile strength and structural scaffold. Rather than a single disease, OI is a spectrum: at one end, a child survives infancy with mild fracture susceptibility and lives a near-normal life; at the other, a baby is born with so many fractures that survival beyond a few hours is impossible. What unites this spectrum is the same fundamental problem: the skeleton's collagen framework is either too scarce, too weak, or structurally disorganized to bear the loads that ordinary movement places on bone.
OI affects approximately 1 in 15,000 to 20,000 live births worldwide, making it one of the more common serious skeletal dysplasias. Because the gene mutations involved are spread across COL1A1, COL1A2, and more than a dozen other genes, the disorder cannot be attributed to a single mutational hot spot. The great majority of cases — roughly 85 to 90 percent — stem from mutations in the two collagen I genes, and these are inherited in an autosomal dominant pattern. That means a single defective copy from one parent is enough to cause the disease. Spontaneous (de novo) mutations account for a meaningful minority of cases, particularly the more severe types, because the mutations responsible for the worst outcomes often reduce reproductive fitness, limiting their transmission across generations.
The Sillence classification, proposed by David Sillence and colleagues in 1979 and subsequently expanded, remains the organizing framework for OI. The original four types — I through IV — map clinical severity from mild (Type I) to perinatal lethal (Type II) to progressively deforming (Type III) to moderate (Type IV). Over the past two decades, advances in molecular genetics have revealed a second, genetically distinct group of OI syndromes caused by mutations in non-collagen genes. These are labeled Types V through XXI in the current expanded classification and together account for the remaining 10 to 15 percent of OI. Despite the expansion, the Sillence types remain clinically useful because phenotype — not just genotype — drives treatment decisions.
Genetics: COL1A1, COL1A2, and Beyond
Type I collagen is the most abundant protein in the human body, forming the structural scaffold for bone, skin, tendons, blood vessel walls, and teeth. Each collagen I molecule is a triple helix made of two alpha-1 chains encoded by COL1A1 and one alpha-2 chain encoded by COL1A2. These chains are synthesized in the endoplasmic reticulum, folded into the triple-helical procollagen molecule, and secreted into the extracellular matrix, where they are cleaved and assembled into collagen fibrils. Any mutation that disrupts the quantity or quality of these chains disrupts the entire downstream process of bone mineralization and fibril formation.
Two broad categories of COL1A1/COL1A2 mutations produce distinct clinical outcomes. The first category is haploinsufficiency: a frameshift, nonsense, or splice-site mutation creates a premature stop codon and the resulting abnormal mRNA is degraded by nonsense-mediated decay. The cell is left producing collagen only from the one normal allele, yielding roughly half the normal amount of structurally correct collagen. This mechanism underlies most cases of Type I OI — the mildest form — because the collagen that is made is qualitatively normal; there is simply less of it. The second and more damaging category involves missense mutations that substitute another amino acid for glycine at specific positions in the collagen triple helix. Every third residue in the helix must be glycine (the smallest amino acid) to fit into the center of the triple-helical structure. A glycine substitution stalls or distorts the folding of the helix, and because the abnormal chain is incorporated alongside the normal chain, a dominant-negative effect poisons the function of the entire molecule. These glycine substitution mutations produce the more severe Types II, III, and IV phenotypes.
The non-collagen OI genes discovered since 2006 encode proteins involved in collagen hydroxylation, chaperoning, and cross-linking — the post-translational modifications required for normal collagen fibril assembly. CRTAP (cartilage-associated protein), LEPRE1 (encoding prolyl 3-hydroxylase 1, P3H1), and PPIB (cyclophilin B) form a complex that hydroxylates a specific proline residue (Pro986) on the alpha-1 chain; loss of any component produces a severe, recessive OI indistinguishable clinically from Type II or III. SERPINH1 encodes HSP47, a collagen-specific chaperone in the endoplasmic reticulum; SERPINF1 encodes PEDF (pigment epithelium-derived factor), a secreted protein important for osteoblast function. IFITM5 is notable because a specific 5'-UTR mutation creates an in-frame start codon that adds five amino acids to the beginning of the protein; this single-nucleotide change accounts for virtually all cases of Type V OI, characterized by hyperplastic callus and a distinctive inter-osseous membrane calcification in the forearm. The collective lesson from these discoveries is that OI is not a single-gene disease but a disease of the collagen I biosynthetic pathway, and any node in that pathway is a potential target for mutation.
Pathophysiology of Collagen Defects
Normal bone strength depends on two complementary components: the mineral phase (hydroxyapatite crystals providing compressive stiffness) and the organic matrix phase (type I collagen fibrils providing tensile flexibility and toughness). In healthy lamellar bone, collagen fibrils are laid down in parallel sheets with alternating fiber directions — a plywood-like architecture that distributes stress and resists crack propagation. In OI bone, the collagen matrix is disorganized. Whether because there is too little collagen (haploinsufficiency) or structurally abnormal collagen (dominant negative), the resulting matrix cannot template normal fibril assembly, and the bone that mineralizes on top of this disorganized scaffold is woven rather than lamellar. Woven bone is weaker, more isotropic, and more brittle — it breaks at loads that lamellar bone handles easily.
The cellular defect in OI is not simply a passive structural weakness. OI osteoblasts actively produce abnormal or insufficient matrix, and in many forms of OI the osteoblasts undergo premature apoptosis after sensing that the protein they are manufacturing is misfolded — a form of endoplasmic reticulum stress. The resulting imbalance between osteoblast and osteoclast activity shifts bone remodeling toward net resorption. Paradoxically, the osteoclasts in OI are often functionally normal; they resorb the existing defective matrix efficiently. This means that the overall effect is: defective matrix is made, some of it is resorbed, and what remains is structurally inferior. Cortical bone is thin, trabecular architecture is sparse, and the periosteum — the fibrous outer sheath of bone that normally contributes to fracture resistance — is thinner than normal.
Understanding this cellular pathophysiology explains why bisphosphonates — drugs that inhibit osteoclast-mediated bone resorption — have become the cornerstone of OI pharmacotherapy. By slowing osteoclastic resorption, bisphosphonates allow even the defective matrix to accumulate over time. Cortices thicken, vertebral body height is partially restored, and bone mineral density (BMD) as measured by dual-energy X-ray absorptiometry (DXA) increases significantly. Crucially, bisphosphonates do not fix the collagen defect — the matrix that accumulates is still qualitatively abnormal — but by increasing its quantity they shift the mechanical balance toward fewer fractures. Studies in children treated with cyclic intravenous pamidronate have shown that vertebral reshaping (partial reversal of compression fractures) occurs in growing bone, a finding that is biologically remarkable because adults cannot achieve this degree of remodeling.
Clinical Features by Type
Type I OI is the mildest and most common form, accounting for roughly half of all cases. Patients have blue sclerae — a characteristic blue-gray discoloration of the whites of the eyes caused by thin sclerae that allow the underlying choroidal pigment to show through. The bones fracture more easily than normal, particularly in childhood, but the fractures are manageable with standard orthopedic care and the fracture rate often declines after puberty when skeletal maturation is complete. Stature is normal or mildly reduced. About half of Type I patients develop premature hearing loss, typically in the second or third decade of life, due to otosclerosis-like changes in the stapedial footplate (the small ossicle that transmits vibration from the eardrum to the inner ear). Dentinogenesis imperfecta — abnormal dentin formation causing amber-colored, translucent, fracture-prone teeth — is present in roughly 40 percent of Type I patients (designated Type IB); those without DI are Type IA. Overall life expectancy in Type I is normal.
Type II OI is the perinatal lethal form and the most severe. Mutations in this type typically produce a dominant-negative glycine substitution at a critical position in the triple helix, or homozygous loss-of-function mutations in recessive OI genes. The hallmarks on prenatal ultrasound or post-mortem radiograph are a crumpled, shortened femur (the classic "telephone receiver" or "accordion" femur), multiple rib fractures producing a beaded appearance on X-ray, and a poorly mineralized skull that deforms on palpation. The cause of death is almost invariably respiratory failure: the severely deformed chest wall cannot expand adequately to support breathing, and lung hypoplasia from intrauterine compression further limits respiratory reserve. Most affected infants are stillborn or die within hours to days of birth; survival beyond a few weeks is exceptional.
Type III is the most severe form compatible with survival and is characterized by progressive deformity. Infants may appear relatively normal at birth but develop progressive bowing of the long bones and vertebral compression fractures over the first years of life. The deformity is driven by the combined effects of repeated fracture, abnormal bone remodeling, and gravity acting on soft, easily deformable bones. Patients typically develop severe short stature (adult height often below 100 cm), scoliosis, and require a wheelchair for mobility. "Popcorn calcifications" — irregular calcific deposits in the cartilaginous growth plates — are a characteristic radiographic finding in the metaphyses of children with Type III and reflect the chaotic endochondral ossification in severely affected bone. Sclerae are gray-blue in childhood and may lighten to near-white in adults. Basilar invagination — upward displacement of the cervical spine through the foramen magnum — is a potentially life-threatening neurological complication of severe OI that requires monitoring by MRI.
Type IV sits between Types I and III in severity. Sclerae are white or near-white, distinguishing it from Types I and II. Fracture rates are moderate — more than Type I but less than Type III — and deformity is mild to moderate. Short stature is common. Dentinogenesis imperfecta is frequent. The newer OI types (V through XXI) are genetically defined; Type V is clinically distinctive for its hyperplastic callus formation (exuberant, tumor-like bone proliferation at fracture sites that can be mistaken for osteosarcoma) and calcification of the interosseous membrane between the radius and ulna, limiting forearm rotation. Other newer types often mimic Type II or Type III clinically but are confirmed by genetic testing.
Extra-Skeletal Manifestations
Blue sclerae are so strongly associated with OI that their presence in a child with unexplained fractures should immediately raise suspicion for the diagnosis. The discoloration arises because the scleral collagen is thinner and less opaque than normal, allowing the blue-black choroidal vascular layer to be visible through the scleral coat. Importantly, blue sclerae do not impair vision and cause no discomfort — they are a sign, not a symptom. However, thin sclerae may increase the theoretical risk of globe rupture from blunt trauma, and ophthalmologic monitoring is reasonable in severely affected patients. Blue sclerae are most prominent in Types I and II; they lighten with age in Types III and IV.
Dentinogenesis imperfecta (DI) is a separate collagen defect affecting the dentin — the layer of mineralized tissue beneath the enamel that makes up the bulk of each tooth. In DI, the dentin is structurally weak, causing the enamel to crack and chip off the dentin surface. The exposed dentin wears rapidly, leading to teeth that are short, discolored (amber to gray-brown), and translucent. Both primary and permanent teeth are affected, though primary teeth tend to show the worst changes. The dental implications are significant: early and regular dental care is essential, and children with DI typically need resin-bonded composite overlays or stainless steel crowns to protect the remaining tooth structure. Teeth that are severely worn or symptomatic may require extraction. It is worth knowing that DI can also occur as an isolated genetic condition (DI types I and II, caused by DSPP mutations) entirely unrelated to OI — so the diagnosis of OI cannot rest on DI alone.
Hearing loss is among the most functionally significant extra-skeletal features for adults living with OI. It affects approximately 50 percent of Type I patients by midlife and is even more prevalent in older adults with Type III. The mechanism is analogous to otosclerosis: abnormal collagen in the ossicular chain of the middle ear leads to progressive fixation of the stapes, the smallest bone in the body, reducing its ability to transmit sound vibration to the cochlea. The result is a conductive or mixed (conductive plus sensorineural) hearing loss that begins in the mid-frequency range and progresses over years. Hearing aids are helpful in the early and moderate stages. Stapedectomy or stapedotomy — surgical procedures that replace or bypass the fixed stapes — can restore hearing in carefully selected patients, analogous to the same procedure used for classical otosclerosis, though the surgical risk may be higher given the fragility of OI bone. A nationwide Finnish study (Kuurila et al., 2002) found that more than half of Finnish adults with OI had clinically significant hearing loss, confirming that this complication deserves routine audiological surveillance starting in adolescence.
Joint hypermobility is common because the same collagen defect that weakens bone also reduces the tensile strength of ligaments and joint capsules. Unlike Ehlers-Danlos syndrome, where hypermobility can be severe and disabling, the hypermobility in OI is generally mild to moderate. Easy bruising from fragile capillaries and mild skin hyperelasticity — detectable on clinical examination but far less prominent than in EDS — round out the connective tissue picture. Patients with OI may also experience chronic musculoskeletal pain disproportionate to the current fracture burden, likely reflecting a combination of micro-damage in the skeleton, muscle weakness from inactivity, and central sensitization — a pattern that deserves assessment and management independent of the structural bone problem.
Diagnosis
In most cases OI is a clinical diagnosis assembled from the combination of recurrent fractures, characteristic physical findings, family history, and imaging. The key clinical triad — fractures with minimal trauma, blue sclerae, and dentinogenesis imperfecta — is sufficiently specific that when all three are present the diagnosis is essentially established. In practice, however, many patients present with only one or two features, particularly in Type I where sclerae may be only faintly blue and DI may be absent. The first priority in any child with unexplained fractures is to distinguish OI from non-accidental injury (child abuse, NAI). This distinction is among the most consequential in all of pediatric medicine: a missed OI diagnosis can result in a family being wrongly accused of abuse, while a missed abuse diagnosis leaves a child at ongoing risk. The features that favor OI over abuse include blue sclerae, wormian bones on skull X-ray, a family history of fractures or blue sclerae, and fractures in locations atypical for abuse (metaphyseal versus diaphyseal, long bones versus ribs in very young infants). A genetics or metabolic bone specialist should be involved whenever OI is on the differential.
Radiographic findings play an important supporting role. Wormian bones — small, irregular ossification centers within the cranial sutures, visible on plain skull X-ray — are highly characteristic of OI, though not pathognomonic; they are found in the majority of patients with Types I, III, and IV. Long bone films show thin cortices, bowing deformities in severe types, and the metaphyseal popcorn calcifications of Type III. Vertebral films may show biconcave or wedge-shaped compression fractures. DXA scanning measures bone mineral density but is not diagnostic on its own because BMD in OI overlaps with the normal range at certain ages; its primary use is to establish a baseline and monitor treatment response.
When clinical assessment is insufficient, two confirmatory tests are available. Skin biopsy with fibroblast culture allows biochemical analysis of the collagen secreted by the patient's connective tissue cells. Electrophoresis of secreted collagen can reveal a quantitative reduction (confirming haploinsufficiency) or an abnormal electrophoretic pattern with slow-migrating bands (confirming a structural defect in the triple helix) — a test that detects approximately 85 to 90 percent of OI mutations. Iliac crest bone biopsy under general anesthesia, once a gold standard, is now less commonly performed but provides direct visualization of the abnormal collagen architecture. In contemporary practice, molecular genetic testing — sequencing of COL1A1 and COL1A2 first, followed by panel testing for other OI genes if negative — is increasingly the confirmation method of choice. A positive molecular result identifies the specific mutation, enabling accurate genetic counseling about recurrence risk and, in some families, prenatal diagnosis.
Treatment and Management
Bisphosphonates are the cornerstone of pharmacological management for OI and represent one of the most robust treatment advances in pediatric bone disease of the past three decades. The landmark 1998 trial by Glorieux and colleagues at Shriners Hospital Montreal demonstrated that cyclic intravenous pamidronate — given as three-day infusions every three to four months in children with severe OI — produced significant increases in vertebral bone mineral density, decreased fracture rates, and, remarkably, partial reshaping of vertebral bodies that had been compressed. Intravenous zoledronic acid, given as a single annual infusion, has largely replaced pamidronate in many centers because it is more convenient and equally effective. For adults and some older children, oral bisphosphonates (alendronate, risedronate) are an alternative, though GI tolerability and adherence can be limiting. The 2016 Cochrane review by Dwan and colleagues, which analyzed all available randomized trials, confirmed that bisphosphonate therapy reliably increases BMD and reduces radiographic fracture rates in OI, with the strongest evidence in children using IV routes. The benefit is genuine but must be placed in perspective: bisphosphonates do not fix the underlying collagen defect, and the fracture rate remains higher than in unaffected peers even on treatment.
Orthopedic surgery is the other major pillar of management, particularly for patients with Types III and IV. The fundamental procedure is intramedullary rodding: a metal rod is inserted through the medullary canal of a deformed long bone (typically the femur or tibia) to straighten the bone and prevent it from re-fracturing and re-deforming in the same crooked orientation. Conventional solid rods require replacement every few years as the child grows, but telescoping (extensible) rods — most notably the Fassier-Duval rod, which expands automatically as the bone lengthens — have substantially reduced the need for reoperations. The timing of rodding is individualized: ideally performed electively, before a bone has re-deformed to the point where sitting, standing, or transfers become impossible, rather than urgently after a comminuted fracture in a severely bent bone. Spinal surgery (posterior spinal fusion for progressive scoliosis) is sometimes required in Type III patients, though it carries higher-than-usual risks given the soft, abnormal bone and the risk of instrument pull-out.
Physical and occupational therapy are essential throughout the lifespan. Aquatherapy (hydrotherapy) is particularly well suited to OI because the buoyancy of water markedly reduces the impact forces on bone, allowing patients to exercise, strengthen muscles, and improve proprioception with far lower fracture risk than land-based exercise. Strong muscles protect bones; immobility accelerates bone loss and deconditioning. The goal of rehabilitation is to maximize function and independence at every age — from assisting infants with safe positioning and handling, to teaching toddlers adaptive movement strategies, to helping adolescents and adults optimize mobility and community participation. Contact sports and high-impact activities are generally avoided, but a thoughtfully individualized approach is preferable to blanket prohibition of all activity. Hearing aids, dental appliances (resin overlays, crowns), and pain management programs round out the multidisciplinary care team for patients with more severe OI.
Emerging Therapies and Research
The most clinically advanced emerging therapy is anti-sclerostin treatment. Sclerostin, encoded by the SOST gene, is a protein secreted by osteocytes (mature bone cells embedded in the mineralized matrix) that inhibits the Wnt signaling pathway and thereby suppresses osteoblast-driven bone formation. Romosozumab, a monoclonal antibody that neutralizes sclerostin, was approved for postmenopausal osteoporosis in 2019 and is now in Phase 2 and Phase 3 trials for OI. The rationale is compelling: instead of merely slowing bone resorption as bisphosphonates do, romosozumab actively stimulates new bone formation — a theoretical advantage in OI, where the problem is not excess resorption but deficient production of adequate matrix. Early trial data are promising, though it remains to be seen whether newly formed matrix in OI patients is sufficiently improved in quality to translate the BMD gains into meaningful fracture reduction.
Denosumab, a RANK-ligand inhibitor that suppresses osteoclast formation and activity, has been explored as an alternative to bisphosphonates in OI patients who cannot tolerate bisphosphonates or who have specific forms of OI with recessive inheritance. A 2020 study by Glorieux, Devogelaer, and colleagues published in Bone reported on denosumab use in OI and showed BMD increases comparable to bisphosphonate therapy. One important caution is the rebound effect: denosumab suppresses bone turnover rapidly but its effect wears off quickly when discontinued, potentially triggering a rebound increase in resorption and fracture risk — a concern that requires careful transition planning if denosumab is stopped.
The most ambitious approaches aim to correct the underlying genetic defect. Gene therapy strategies under investigation include antisense oligonucleotides (ASOs) designed to selectively silence the dominant-negative mutant allele while leaving the normal allele intact — in theory converting a severe dominant-negative phenotype into the milder haploinsufficiency phenotype of Type I. Stem cell transplantation, specifically mesenchymal stem cell (MSC) infusion, has been explored in a small number of pediatric patients; the rationale is that donor MSCs engraft in bone as functional osteoblasts and contribute normal collagen to the matrix. Results to date have been modest — the engraftment efficiency in bone is low — but the approach remains biologically compelling. CRISPR-based gene correction is being developed in preclinical OI mouse models. These genetic approaches face significant hurdles in delivery, safety, and efficiency, but given that current therapies all work around the collagen defect rather than fixing it, gene-level correction remains the long-term goal of the OI research community.
Key Research Papers
- Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387(10028):1657–1671. PMID 26542481
- Marini JC, Forlino A, Bächinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. — Search PubMed
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101–116. PMID 458828
- Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339(14):947–952. PMID 9753709
- Trejo P, Rauch F. Osteogenesis imperfecta in children and adolescents — new developments in diagnosis and treatment. Osteoporos Int. 2016;27(12):3427–3437. — Search PubMed
- Lim J, Grafe I, Alexander S, Lee B. Genetic causes and mechanisms of osteogenesis imperfecta. Bone. 2017;102:40–48. — Search PubMed
- Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377–1385. PMID 15110498
- Bishop N, Adami S, Ahmed SF, et al. Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9902):1424–1432. PMID 23927913
- Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2016;10:CD005088. PMID 27760454
- Semler O, Garbes L, Keupp K, et al. A mutation in the 5'-UTR of IFITM5 creates an in-frame start codon and causes atypically severe osteogenesis imperfecta type V. Am J Hum Genet. 2012;91(2):349–357. — Search PubMed
- Glorieux FH, Devogelaer JP, Brochhausen C, et al. Denosumab for treatment of osteogenesis imperfecta. Bone. 2020;141:115579. — Search PubMed
- Kuurila K, Grénman R, Johansson R, Kaitila I. Hearing loss in Finnish adults with osteogenesis imperfecta: a nationwide survey. Ann Otol Rhinol Laryngol. 2002;111(10):939–946. — Search PubMed
PubMed topic searches:
Osteogenesis imperfecta bisphosphonate treatment •
OI collagen mutation •
OI gene therapy •
OI intramedullary rodding surgery
Connections
- Genetics
- Ehlers-Danlos Syndrome
- Marfan Syndrome
- Loeys-Dietz Syndrome
- Calcium
- Vitamin D3
- Phosphorus
- Orthopedic Conditions