Homocystinuria
Overview and Genetics
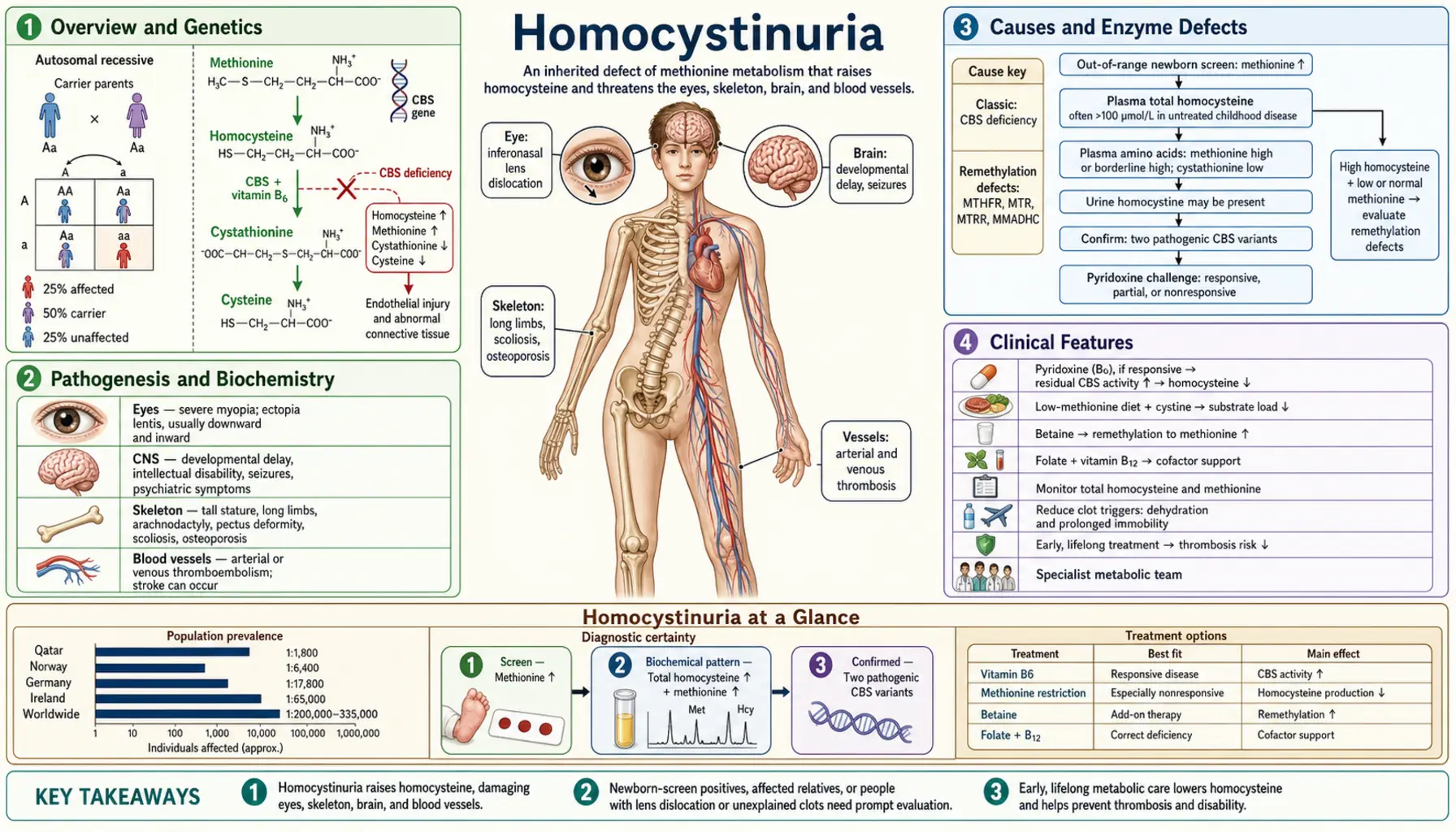
Homocystinuria is a group of inherited metabolic disorders characterized by elevated homocysteine in the blood (homocysteinemia) and its excretion in urine. The most common and clinically important form results from deficiency of cystathionine beta-synthase (CBS), encoded by the CBS gene on chromosome 21q22.3. CBS-deficiency homocystinuria (HCU) affects approximately 1 in 200,000–335,000 newborns worldwide, though some populations have higher prevalence — Ireland has one of the highest rates, estimated at 1 in 65,000.
In normal methionine metabolism, dietary methionine is converted to S-adenosylmethionine (SAM), which donates methyl groups throughout the body. The resulting S-adenosylhomocysteine is hydrolyzed to homocysteine, which at this branch point can either be remethylated back to methionine (using methyltetrahydrofolate + B12, via methionine synthase) or transsulfurated to cystathionine (via CBS using pyridoxal phosphate/B6, then to cysteine). When CBS is deficient, homocysteine cannot enter the transsulfuration pathway and accumulates dramatically — plasma total homocysteine can reach 200–400 µmol/L (normal <15 µmol/L).
Pathogenesis and Biochemistry
Excess homocysteine is toxic to multiple organ systems through several mechanisms:
- Endothelial dysfunction: Homocysteine directly injures vascular endothelium, impairs nitric oxide bioavailability, promotes oxidative stress, and activates the coagulation cascade — the mechanistic basis for the severe thrombophilia seen in HCU.
- Connective tissue disruption: Homocysteine interferes with fibrillin-1 crosslinking and collagen maturation, leading to the marfanoid skeletal habitus, ectopia lentis, and osteoporosis. Homocysteine also inhibits lysyl oxidase, the enzyme that crosslinks collagen and elastin fibers.
- Neurotoxicity: Elevated homocysteine acts as an excitotoxin at NMDA receptors and impairs remethylation of neuronal SAM, reducing availability of methylation substrates for neurotransmitter synthesis and DNA methylation.
- Methionine excess: Because homocysteine accumulates upstream, methionine also builds up — plasma methionine is elevated in CBS-HCU, which itself may contribute to toxicity and is the metabolite detected on newborn screening.
- Cysteine deficiency: Since cystathionine and cysteine cannot be synthesized from the blocked pathway, cysteine becomes conditionally essential — dietary supplementation partially corrects this deficit.
Causes and Enzyme Defects
Several distinct enzyme defects can cause elevated urine homocystine, and they are clinically and biochemically different:
- CBS deficiency (classic HCU): Most common cause. Autosomal recessive. >200 pathogenic CBS variants identified. Produces severe hyperhomocysteinemia with elevated methionine. Responds to pyridoxine (B6) in ~50% of cases.
- MTHFR severe deficiency: Methylenetetrahydrofolate reductase deficiency (distinct from the common C677T/A1298C SNPs, which cause only mildly elevated homocysteine). Severe MTHFR deficiency impairs remethylation of homocysteine back to methionine. In contrast to CBS-HCU, methionine is LOW (not elevated) because the remethylation route is blocked. Presents with neurological features and lower thrombotic risk.
- Cobalamin/B12 pathway defects (cblC, cblD, cblE, cblG): Methylcobalamin cofactor is required for methionine synthase. Defects in cobalamin metabolism impair remethylation, causing combined homocystinuria and methylmalonic acidemia (cblC, cblD) or isolated homocystinuria (cblE, cblG). These disorders present with megaloblastic anemia and neurological disease and respond to hydroxocobalamin treatment.
This review focuses on CBS-deficiency HCU, which accounts for the vast majority of classical homocystinuria presentations.
Clinical Features
CBS-HCU is a multi-system disorder affecting the eyes, skeleton, cardiovascular system, and brain. Untreated individuals appear normal at birth and develop clinical features over the first years of life:
Ocular Features- Ectopia lentis (subluxed lens): Presents in the first decade; occurs in ~90% of untreated patients. The lens dislocates inferiorly and medially — a critical distinguishing feature from Marfan syndrome, where the lens typically dislocates superiorly. Mechanism: homocysteine disrupts fibrillin-1 microfibrils of the suspensory ligaments (zonules of Zinn).
- High myopia: Often severe (>10 diopters), present before or concurrent with lens dislocation. Frequently the first feature noted by ophthalmologists.
- Glaucoma and retinal detachment as secondary complications of untreated lens dislocation.
- Marfanoid habitus: Tall stature, long limbs (dolichostenomelia), arachnodactyly, scoliosis, high-arched palate, pectus excavatum or carinatum. Clinically indistinguishable from Marfan syndrome at first glance.
- Osteoporosis: Vertebral compression fractures can occur in the second decade. Caused by impaired collagen crosslinking and cysteine deficiency (cysteine needed for collagen synthesis).
- Genu valgum (knock-knees) is more common in HCU than in Marfan syndrome.
- Intellectual disability: Present in approximately 50% of untreated CBS-HCU patients. IQ decline correlates with disease control. Early dietary treatment largely prevents cognitive impairment.
- Seizures in ~20% of untreated patients.
- Psychiatric features: depression, anxiety, psychosis, and obsessive-compulsive disorder are more common in CBS-HCU patients than the general population.
- Extrapyramidal movements and focal neurological signs can result from cerebrovascular thrombosis.
- Malar flush: Distinctive rosy/red cheeks, particularly marked in fair-skinned patients.
- Fair phenotype: Light skin, hair, and eyes — cysteine competes with tyrosine for melanin synthesis, and cysteine deficiency shifts melanin production toward the lighter phaeomelanin pathway.
- Livedo reticularis (mottled skin pattern from small vessel involvement).
Homocystinuria vs. Marfan Syndrome
The marfanoid appearance of HCU patients has historically led to diagnostic confusion. Key distinguishing features:
- Lens dislocation direction: HCU → inferior; Marfan → superior (upward). This is one of the most tested clinical distinctions in genetics and ophthalmology.
- Intellectual ability: Most Marfan patients have normal intelligence; ~50% of untreated HCU patients have cognitive impairment.
- Thrombosis: Major feature of HCU; not a primary feature of Marfan syndrome.
- Cardiac: Marfan causes aortic root dilation, mitral valve prolapse; these are absent or mild in HCU.
- Laboratory: HCU shows elevated plasma homocysteine and methionine; Marfan has normal amino acid levels but may have FBN1 mutation on gene panel.
- Osteoporosis: Present and early in HCU; not a defining feature of Marfan.
- Response to B6: Unique to HCU — B6-responsive patients can show dramatic improvement on pyridoxine.
Thromboembolism Risk
Thromboembolic complications are the leading cause of premature death in untreated CBS-HCU. The risk is profound:
- By age 30, approximately 50% of untreated CBS-HCU patients have had a thromboembolic event.
- Events occur at any age — thrombotic strokes in toddlers and DVT/PE in teenagers are documented.
- Both venous (DVT, PE, dural sinus thrombosis) and arterial (coronary, cerebral, renal, peripheral) events occur with unusual frequency and severity for the patient's age.
- Surgical and anesthetic procedures dramatically elevate risk — elective operations require specific HCU thromboprophylaxis protocols.
- Mechanisms include endothelial injury, platelet hyperactivation, impaired fibrinolysis, and acquired protein C/S deficiency in some patients.
- Treatment with methionine-restricted diet, betaine, and/or pyridoxine (where responsive) substantially reduces thrombotic risk alongside appropriate anticoagulation planning for procedures.
- Low-dose aspirin is commonly used; anticoagulation decisions are individualized based on prior events.
Newborn Screening and Diagnosis
CBS-HCU is included in expanded newborn screening programs in most developed countries, though coverage varies:
- Newborn screening marker: Elevated plasma methionine detected by tandem mass spectrometry (MS/MS) on dried blood spot (Guthrie card). Methionine is elevated in CBS-HCU because homocysteine accumulation is upstream of methionine. Important caveat: some mild or B6-responsive CBS-HCU cases have normal methionine at newborn screening — up to 30% of B6-responsive cases may be missed by MS/MS newborn screening alone.
- Confirmatory testing: Plasma amino acid quantitation (elevated methionine, low cystine) + plasma total homocysteine (tHcy) — markedly elevated in CBS-HCU. Urine amino acids show homocystine (the oxidized disulfide dimer of homocysteine).
- Cyanide-nitroprusside test: Historical colorimetric urine test; homocystine gives a positive result (orange/red color). Now largely replaced by quantitative plasma tHcy measurement, but still referenced in classic descriptions.
- Molecular confirmation: CBS gene sequencing identifies the specific pathogenic variant(s) and predicts B6-responsiveness in some cases (though functional testing with a pyridoxine trial is still required clinically).
- Ophthalmological evaluation: Slit-lamp examination for lens dislocation, which may precede diagnosis in late-detected or screening-missed cases.
- Bone density: DXA scan for baseline osteoporosis assessment at diagnosis and periodically thereafter.
Treatment and Management
The goal of treatment is to reduce plasma total homocysteine to safe levels (ideally tHcy <50 µmol/L, with some centers targeting <100 µmol/L for older patients). Multiple complementary strategies exist:
1. Pyridoxine (Vitamin B6) TrialAll CBS-HCU patients should undergo a pyridoxine responsiveness trial at diagnosis. Approximately 50% of patients with CBS-HCU show significant response — tHcy drops to near-normal or normal with pharmacological B6 doses (typically 100–500 mg/day). B6-responsive patients generally have a milder natural history, better prognosis, and may not require strict dietary restriction. B6 acts as cofactor stabilizer for mutant CBS enzyme. Non-responsive patients require full dietary therapy.
2. Methionine-Restricted DietFor B6-non-responsive patients, a low-methionine diet is the cornerstone of treatment. Since methionine is abundant in protein-containing foods, the diet requires restriction of natural protein and substitution with a methionine-free amino acid formula. This is demanding — similar in rigor to the phenylalanine-restricted diet in PKU. Cystine (normally produced via transsulfuration) must be supplemented as it becomes conditionally essential.
3. Betaine (Trimethylglycine)Betaine (available as Cystadane) is an alternative methyl donor that promotes remethylation of homocysteine to methionine via a B12-independent pathway (betaine-homocysteine methyltransferase, BHMT). This bypasses the blocked CBS enzyme by recycling homocysteine back to methionine. Betaine is effective in both B6-responsive and non-responsive patients and is particularly useful in patients who cannot achieve adequate dietary control. Caution: because betaine drives methionine formation, methionine can rise further — cerebral edema has been reported rarely with very high methionine levels; monitoring is essential.
4. Folate and B12 SupplementationOptimal B12 (cobalamin) and folate levels maximize methionine synthase activity in the remethylation pathway, providing an additional route for homocysteine disposal. These should be maintained at the upper end of normal in all HCU patients.
5. Antithrombotic TherapyLow-dose aspirin is widely used. Perioperative anticoagulation protocols are mandatory for elective procedures. Patients with prior thrombotic events typically require long-term anticoagulation. Some centers use prophylactic anticoagulation for high-risk procedures or travel (e.g., long-haul flights).
6. MonitoringRegular plasma tHcy, amino acid profiles, ophthalmological reviews, bone density, and neurodevelopmental assessment form the monitoring backbone. Annual or biannual frequency depending on disease control and patient age.
Pyridoxine-Responsive vs. Non-Responsive HCU
The distinction between B6-responsive and B6-non-responsive CBS-HCU is clinically crucial because it dramatically changes the treatment plan, prognosis, and quality of life:
- B6-responsive (~50% of CBS-HCU): Generally have milder CBS mutations that leave some residual enzyme activity; pharmacological B6 raises cofactor levels enough to boost mutant enzyme activity significantly. These patients can often be managed with B6 alone (plus folate/B12), without the burdensome low-methionine diet. Natural history is substantially better: lower rates of intellectual disability, fewer thromboembolic events, milder ocular disease.
- B6-non-responsive (~50%): Have little or no residual CBS activity. Require full methionine restriction + amino acid formula + betaine. More demanding to manage; historically worse outcomes if dietary control is suboptimal.
- Responsiveness testing: A formal trial of high-dose pyridoxine (100–500 mg/day for 3–6 weeks) with pre/post plasma tHcy measurement. A >30–50% decrease in tHcy, or normalization, defines B6-responsiveness. Some experts recommend trialing in infancy before committing to dietary restrictions.
- Some CBS variants are partially responsive — meaningful but incomplete tHcy reduction. These patients benefit from combined pyridoxine + dietary restriction or betaine.
- Molecular genotype can suggest likely responsiveness (certain missense variants are strongly associated with B6-responsiveness) but cannot replace functional testing.
Prognosis and Long-Term Outcomes
With early diagnosis via newborn screening and appropriate treatment, most CBS-HCU patients can expect near-normal life outcomes. Specific findings from long-term studies:
- Early-treated B6-responsive patients: intellectual outcomes essentially normal; thrombotic events rare with maintained tHcy control.
- Early-treated B6-non-responsive patients on strict dietary control: significant reduction (but not elimination) of complications compared to untreated historical cohorts.
- Late-diagnosed patients (pre-newborn screening era): average IQ around 60–80 in non-responsive individuals; 50% had a thromboembolic event by age 30; ~25% had ectopia lentis by age 5, ~80% by age 15.
- The International HCU Network (HCUnetwork.org) has published long-term natural history data from registries across multiple countries showing that metabolic control, as reflected by tHcy, is the strongest predictor of outcomes.
- Osteoporosis remains a long-term concern even in treated patients and requires DXA monitoring, calcium, vitamin D, and weight-bearing exercise.
- Psychiatric and behavioral complications may persist despite good metabolic control and benefit from mental health support.
Key Research Papers
- Mudd SH, Skovby F, Levy HL, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985;37(1):1–31. — Search PubMed — Landmark retrospective study of 629 patients; defined the spectrum of untreated HCU complications.
- Yap S, Naughten E. Homocystinuria due to cystathionine beta-synthase deficiency in Ireland: 25 years' experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J Inherit Metab Dis. 1998;21(7):738–747. — Search PubMed — Irish cohort; demonstrates improved outcomes with early screening and treatment.
- Stabler SP, Lindenbaum J, Savage DG, Allen RH. Elevation of serum cystathionine levels in patients with cobalamin and folate deficiency. Blood. 1993;81(12):3404–3413. — Search PubMed — Key paper establishing B12/folate pathway interplay with homocysteine metabolism.
- Wilcken DE, Wilcken B. The pathogenesis of coronary artery disease: a possible role for methionine metabolism. J Clin Invest. 1976;57(4):1079–1082. — Search PubMed — Early paper linking homocysteine to cardiovascular disease; foundational for understanding thrombosis in HCU.
- Whiteman P, Clayton PT, Krywawych S, Morley K, Rumsby G. Homocystinuria diagnosed by urinary amino acid analysis. Ann Clin Biochem. 2006;43(Pt 2):170–172. — Search PubMed — Biochemical diagnostic approach and laboratory detection of urine homocystine.
- Skovby F, Gaustadnes M, Mudd SH. A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol Genet Metab. 2010;99(1):1–3. — Search PubMed — Update to Mudd 1985 with modern biochemical monitoring data.
- Naughten ER, Yap S, Mayne PD. Newborn screening for homocystinuria: Irish and world experience. Eur J Pediatr. 1998;157(Suppl 2):S84–S87. — Search PubMed — Comparative newborn screening data across countries.
- Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically. Arterioscler Thromb Vasc Biol. 2001;21(12):2080–2085. — Search PubMed — Quantitative thrombosis risk reduction with treatment; major outcome study.
- Morris AA, Kozich V, Santra S, et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017;40(1):49–74. PMID 27778219 — Current European clinical guidelines for CBS-HCU diagnosis and management.
- Singh RH, Whitehead TM. Genetic metabolic dietetics. In: Nutrition in the Treatment of Genetic Diseases; methionine-restricted diet protocol and amino acid formula use in HCU management. PubMed search: homocystinuria methionine restricted diet
- Wilcken B, Turner G. Homocystinuria in New South Wales. Arch Dis Child. 1978;53(4):242–245. — Search PubMed — Early newborn screening cohort; documents benefit of early detection.
- Schwahn BC, Hafner D, Hohlfeld T, Zschocke J, Lindner M, Schadewaldt P, Wendel U. Pharmacokinetics of oral betaine in healthy subjects and patients with homocystinuria. Br J Clin Pharmacol. 2003;55(1):6–13. — Search PubMed — Betaine pharmacokinetics and dosing rationale in HCU treatment.
Search PubMed: homocystinuria CBS cystathionine beta-synthase
Search PubMed: homocystinuria thromboembolism treatment
Search PubMed: pyridoxine responsive homocystinuria
Connections
- Genetics
- Phenylketonuria (PKU)
- Maple Syrup Urine Disease
- Marfan Syndrome
- Vitamin B6 (Pyridoxine)
- Vitamin B12 (Cobalamin)
- Folate (Vitamin B9)
- Methionine
- Cysteine
- Thrombosis
- Homocysteine Lab Test
- Eye Diseases