Maple Syrup Urine Disease
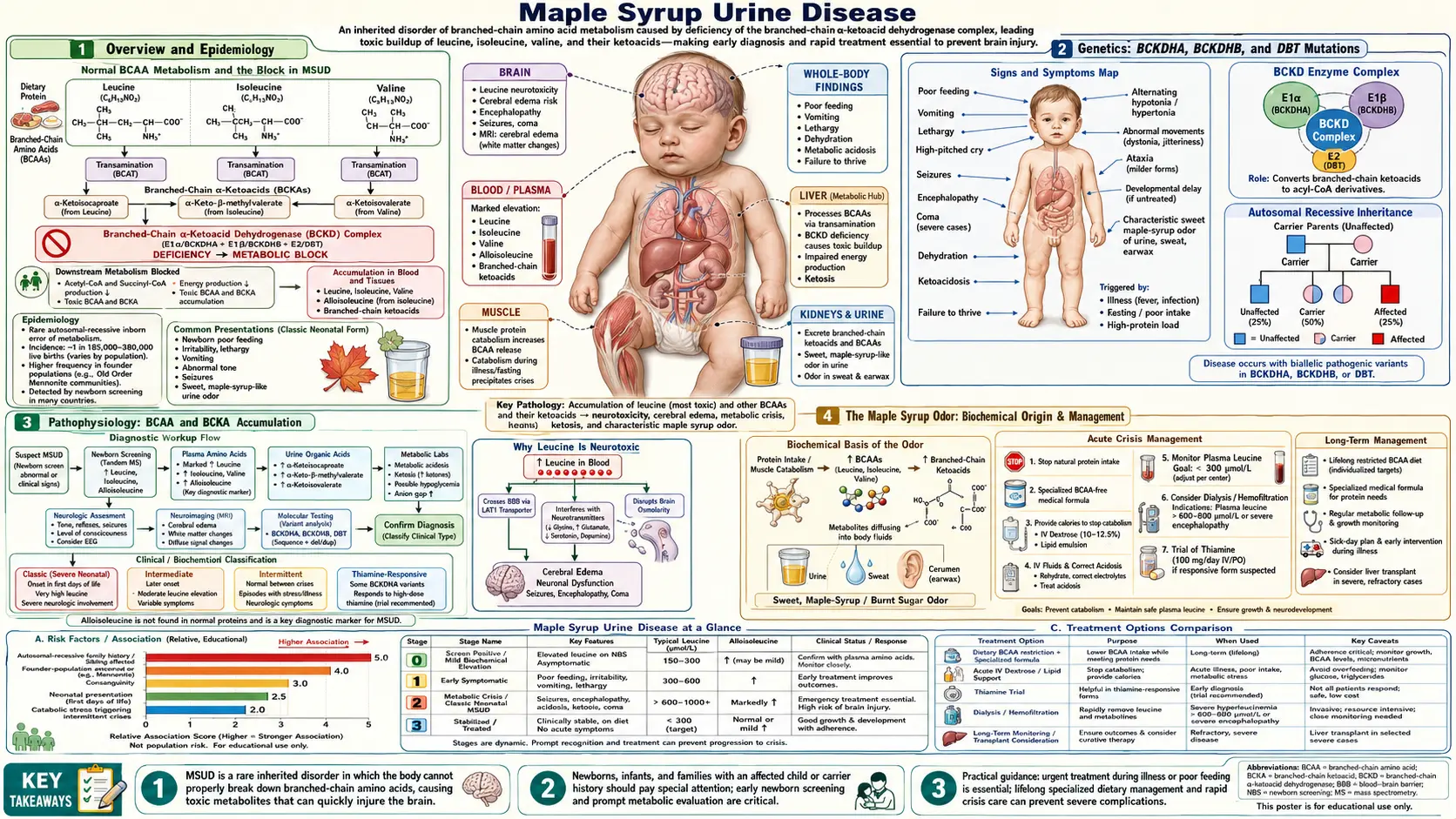
Overview and Epidemiology
Maple Syrup Urine Disease (MSUD) is an autosomal recessive inborn error of branched-chain amino acid (BCAA) catabolism caused by deficiency of the branched-chain alpha-keto acid dehydrogenase (BCKAD) complex. The three BCAAs — leucine, isoleucine, and valine — accumulate in blood and tissue along with their respective branched-chain keto acid (BCKA) metabolites. Without treatment, classic MSUD presents as a neonatal neurological emergency leading to cerebral edema and death.
The disease is named for the characteristic sweet maple syrup or caramel odor of urine, sweat, and earwax caused by accumulating BCKA metabolites. First described by Menkes et al. in 1954 in four siblings who died of a rapidly progressive neurological illness in infancy, MSUD is now included on the Recommended Uniform Screening Panel (RUSP) for U.S. newborn screening programs.
Prevalence: approximately 1:185,000 live births in the general population. Dramatically higher in Old Order Mennonite and Amish communities — approximately 1:380 live births — due to a founder mutation (Y393N in the BCKDHA gene). More than 200 pathogenic variants across three causal genes have been documented worldwide.
Genetics: BCKDHA, BCKDHB, and DBT Mutations
The BCKAD complex is a mitochondrial multi-enzyme complex with four subunit types. Three genes account for the vast majority of MSUD cases:
- BCKDHA (chromosome 19q13.2): encodes the E1α subunit. This is the most commonly mutated gene. The Mennonite founder mutation Y393N resides here and accounts for a large proportion of MSUD cases in Old Order Mennonite and Amish populations.
- BCKDHB (chromosome 6q14.1): encodes the E1β subunit. Mutations here are the second most common cause of MSUD.
- DBT (chromosome 1p21.2): encodes the E2 subunit (dihydrolipoyl transacylase). Thiamine-responsive forms of MSUD are often associated with DBT mutations.
A fourth subunit, E3 (dihydrolipoamide dehydrogenase, encoded by the DLD gene on chromosome 7q31.1), is shared with the pyruvate dehydrogenase complex and the alpha-ketoglutarate dehydrogenase complex. E3 deficiency therefore causes a combined organic acidemia affecting all three complexes simultaneously, producing a more severe and metabolically complex phenotype.
MSUD follows strict autosomal recessive inheritance. Residual BCKAD enzyme activity correlates directly with clinical severity: less than 2% residual activity produces classic MSUD; 3–30% produces the intermediate form; greater than 30% with stress-triggered crises produces the intermittent subtype. More than 200 pathogenic variants have been documented across the three main genes.
Pathophysiology: BCAA and BCKA Accumulation
Under normal physiology, dietary leucine, isoleucine, and valine are first transaminated by the branched-chain aminotransferase (BCAT) enzyme — predominantly in muscle and brain — to produce their respective branched-chain keto acids (BCKAs): alpha-ketoisocaproic acid (KIC) from leucine, alpha-keto-beta-methylvaleric acid (KMV) from isoleucine, and alpha-ketoisovaleric acid (KIV) from valine. These BCKAs then undergo oxidative decarboxylation by the BCKAD complex to yield acyl-CoA intermediates that enter the TCA cycle for energy production.
In MSUD, the BCKAD complex is deficient. BCKAs cannot be decarboxylated and therefore accumulate in plasma, urine, and tissues alongside their parent BCAAs.
Leucine and KIC are the most neurotoxic species:
- Leucine competes at the LAT1 large neutral amino acid transporter at the blood-brain barrier, displacing tyrosine, tryptophan, and phenylalanine. This reduces brain uptake of these precursors and impairs synthesis of dopamine, serotonin, and norepinephrine.
- KIC directly inhibits glutamate dehydrogenase, disrupting the balance of GABA and glutamate neurotransmitters.
- Leucine and KIC cause cerebral edema through intracellular mechanisms — astrocyte swelling is particularly prominent — contributing to the neonatal encephalopathy.
- Both leucine and KIC impair mitochondrial function, causing neuronal energy failure that accelerates the cascade toward irreversible brain injury.
A diagnostically important byproduct of the blocked pathway is alloisoleucine, which forms non-enzymatically from the accumulated KMV (2-oxo-3-methylvalerate). Alloisoleucine is not normally present in human plasma, and its detection on quantitative amino acid analysis is considered pathognomonic for MSUD.
The Maple Syrup Odor: Biochemical Origin
The hallmark sweet, caramel-like maple syrup odor that gives the disease its name arises from the accumulation of branched-chain keto acids, particularly through the production of sotolone (4,5-dimethyl-3-hydroxy-2(3H)-furanone), a lactone metabolite derived from the isoleucine keto acid pathway. Sotolone is also the primary compound responsible for the scent of fenugreek seeds and artificial maple flavoring used in food products.
The odor is detectable in urine, in cerumen (earwax — often the first location where parents or nurses notice it), and in sweat. In untreated classic MSUD, the odor is present from birth, well before the neurological crisis becomes apparent. It can also be detected by clinicians examining diapers during a metabolic crisis.
Before the era of tandem mass spectrometry newborn screening, the maple syrup odor served as a critical early diagnostic clue that prompted urgent investigation. Even today, the odor in an acutely ill neonate should trigger immediate plasma amino acid testing. Parents of diagnosed children are counseled to recognize changes in the intensity of the odor as an early warning sign of metabolic decompensation during illness.
MSUD Subtypes: Classic, Intermediate, Intermittent, Thiamine-Responsive, and E3-Deficient
MSUD is not a single uniform disorder. Five clinically and biochemically distinct subtypes are recognized, each associated with different levels of residual BCKAD enzyme activity and different clinical trajectories.
Classic MSUD
The most common and most severe subtype. Residual BCKAD activity is less than 2% of normal. Presents with neonatal metabolic crisis at 3–5 days of life. The maple syrup odor is present from birth. All three BCAAs are markedly elevated, and alloisoleucine is invariably detected on plasma amino acid analysis. Without urgent treatment, classic MSUD is fatal in the newborn period.
Intermediate MSUD
Residual BCKAD activity is 3–30% of normal. Patients do not experience acute neonatal crises but have chronic elevation of BCAAs leading to intellectual disability, poor growth, and developmental delay presenting during infancy or childhood. The diagnosis is often made later than classic MSUD.
Intermittent MSUD
Residual BCKAD activity is 5–15% of normal — sufficient for normal BCAA metabolism under basal conditions. Patients are asymptomatic and develop normally between episodes. However, catabolic stress from intercurrent infection, surgery, fasting, or excessive protein intake can trigger an acute crisis clinically indistinguishable from classic MSUD. Diagnosis is frequently delayed because routine newborn screening may miss patients with this subtype; the first identified crisis may occur in older childhood.
Thiamine-Responsive MSUD
Often associated with DBT or BCKDHA mutations. High-dose thiamine supplementation (50–300 mg/day) improves residual BCKAD complex activity, leading to measurable reductions in plasma leucine, isoleucine, and valine levels. Clinical response is variable and does not eliminate the need for dietary management entirely in most patients. A thiamine trial should be attempted in all newly diagnosed MSUD patients to identify responders.
E3-Deficient MSUD (Combined)
Caused by mutations in the DLD gene encoding the E3 (dihydrolipoamide dehydrogenase) subunit, which is shared by three mitochondrial enzyme complexes. E3 deficiency therefore simultaneously impairs the BCKAD complex, the pyruvate dehydrogenase complex, and the alpha-ketoglutarate dehydrogenase complex. The result is a combined organic acidemia with features of MSUD plus lactic acidosis and alpha-ketoglutaric aciduria. This subtype is often severe and metabolically complex to manage, requiring attention to multiple metabolic pathways simultaneously.
Clinical Presentation: Neonatal Crisis and Long-Term Complications
The timeline of the classic neonatal presentation is characteristic and closely tied to the accumulation of dietary leucine after feeding is established:
- Days 1–2: apparently normal. Colostrum and early breast milk contain relatively low leucine compared with formula, so leucine loading begins gradually after feeding is well established.
- Days 3–4: poor feeding, irritability, lethargy — early nonspecific signs that may be dismissed as normal newborn behavior.
- Days 4–5: progressive encephalopathy — stupor transitioning to coma, alternating hypotonia and rigidity, stereotyped movement patterns including "bicycling" or "fencing" dystonic posturing, opisthotonus, and a bulging fontanelle signaling rising intracranial pressure.
- Days 5–7 if untreated: brainstem herniation, respiratory failure, and death.
The maple syrup odor is detectable throughout the neonatal period in classic disease.
MRI findings in acute MSUD are nearly pathognomonic: cytotoxic edema with diffusion-weighted imaging (DWI) restricted diffusion preferentially affecting the deep brain structures — cerebellar white matter, brainstem, cerebral peduncles, posterior limb of the internal capsule, and globus pallidus. This distribution mirrors the myelination pattern of the newborn brain, because myelinated regions accumulate BCAA metabolites preferentially. This distinctive MRI appearance strongly suggests MSUD even before biochemical confirmation.
Long-term complications in treated survivors include:
- Intellectual disability — severity correlates with the duration and intensity of the neonatal crisis and with the quality of subsequent metabolic control throughout childhood.
- Spastic quadriplegia following severe neonatal encephalopathy.
- Seizures, often refractory to standard antiepileptic treatment.
- Psychiatric disorders — depression, anxiety, and ADHD occur at elevated rates even in patients with excellent metabolic control, suggesting that chronic low-level BCAA dysregulation or neurotransmitter imbalance contributes independently of acute crises.
- Recurrent pancreatitis — mechanism is not fully established.
- Metabolic crises throughout life, triggered by any catabolic state such as infection, fever, surgery, or prolonged fasting.
Diagnosis: Newborn Screening and Confirmatory Testing
Newborn screening in the United States uses tandem mass spectrometry (MS/MS) on dried blood spots collected at 24–48 hours of age. Key markers include:
- Elevated leucine plus isoleucine (these two amino acids co-elute at the same mass of 132 Da and cannot be distinguished from each other by standard MS/MS).
- Elevated valine.
- Detection of alloisoleucine when present at detectable levels — pathognomonic.
- Elevated leucine/(isoleucine + valine) ratio.
- Elevated leucine/phenylalanine ratio to reduce false positives.
Sensitivity for classic MSUD exceeds 99%. Intermittent MSUD may be missed if sampled during a period of normal BCAA levels.
Confirmatory testing includes:
- Quantitative plasma amino acids: markedly elevated leucine, isoleucine, and valine. The presence of alloisoleucine (greater than 5 μmol/L) is pathognomonic — this amino acid is not normally present in human plasma at detectable levels.
- Urine organic acids: elevated BCKA metabolites including 2-oxoisocaproic acid (from leucine), 2-oxo-3-methylvaleric acid (from isoleucine), and 2-oxoisovaleric acid (from valine).
- BCKAD enzyme activity measured in leukocytes or cultured skin fibroblasts — quantifies residual activity and helps classify the subtype.
- Molecular gene sequencing of BCKDHA, BCKDHB, and DBT — confirms diagnosis, identifies the specific mutation for family studies, and helps predict thiamine responsiveness.
Monitoring during treatment: plasma BCAAs via dried blood spot cards at home. Target plasma leucine is 75–300 μmol/L during treatment (normal less than 200 μmol/L). Leucine is the primary target for monitoring and dietary adjustments.
Treatment: Acute Crisis Management and Chronic Dietary Therapy
The fundamental goal of acute treatment is to rapidly reduce plasma leucine — the most neurotoxic BCAA — while simultaneously maintaining anabolism to prevent the breakdown of endogenous protein that would release additional leucine from muscle.
Acute Crisis Management
- High-rate intravenous 10% dextrose at a glucose infusion rate of 8–10 mg/kg/min or higher, combined with insulin at 0.05–0.1 units/kg/hour. This combination drives a strongly anabolic state, shifting BCAAs into protein synthesis and causing rapid reduction of plasma leucine, isoleucine, and valine.
- Stop all exogenous BCAA intake — hold protein intake and BCAA-containing formula for 24–48 hours during the acute phase.
- BCAA-free amino acid solution administered intravenously or orally. This provides all essential amino acids minus leucine, isoleucine, and valine, preventing catabolism of endogenous protein stores and accelerating the leucine reduction.
- Dialysis — hemodialysis is preferred over peritoneal dialysis for speed of effect — when plasma leucine exceeds 1,500–2,000 μmol/L, or when neurological deterioration continues despite maximal medical management. Dialysis is the most rapid method available for reducing plasma leucine.
- Plasma amino acids should be monitored every 4–6 hours during the crisis. Dietary protein is not reintroduced until plasma leucine falls below 300 μmol/L.
- Thiamine 100–300 mg/day should be started in all patients presenting with MSUD while awaiting determination of thiamine responsiveness.
Chronic Dietary Management
Long-term treatment is built around a specialized medical formula that provides all essential amino acids, calories, vitamins, and minerals while omitting leucine, isoleucine, and valine. Small, carefully measured amounts of natural protein are added — titrated to plasma leucine levels and the patient's growth requirements. Typical leucine intake in infancy is approximately 40–80 mg of leucine per day from natural protein, increasing with age and adjusted based on home dried blood spot monitoring results.
Frequent monitoring is essential: dried blood spot testing 2–3 times per week in infancy and weekly in older patients during stable periods. All patients and families must have an emergency sick-day protocol — increasing formula, reducing natural protein, and contacting the metabolic team at the first sign of intercurrent illness or catabolism. A missed meal or a febrile illness can trigger a life-threatening crisis within hours in classic and intermediate MSUD.
Liver Transplantation in MSUD
The rationale for liver transplantation in MSUD rests on the fact that the BCKAD enzyme complex is expressed predominantly in hepatic tissue. A successful liver transplant restores approximately 85–90% of total body BCKAD enzyme activity and is essentially curative with respect to metabolic crises.
First reported in 2006, subsequent case series have consistently demonstrated that liver transplantation in classic MSUD resolves recurrent metabolic crises, allows near-normal dietary intake including natural protein, and substantially improves quality of life. More than 95% of transplanted patients experience zero metabolic crises post-transplant. Plasma leucine, isoleucine, and valine normalize on a regular diet in the majority of recipients.
Liver transplantation does not reverse pre-existing neurological damage — intellectual disability and other deficits acquired before or during the procedure are not recovered. The best outcomes are achieved when transplantation is performed early, before irreversible injury accumulates from repeated crises or chronic BCAA elevation.
A particularly important consideration is that heterozygous donor livers — from carriers with one normal and one pathogenic allele — provide sufficient BCKAD activity (approximately 50% of normal, which far exceeds the threshold needed to handle a normal diet with no crisis risk). This has made living-related liver donation feasible in Mennonite and Amish communities where carriers are common, allowing donors to contribute to multiple family members over time with relatively lower surgical risk than obtaining cadaveric organs.
Transplantation is increasingly considered standard of care for classic MSUD, particularly where the burden of dietary management and metabolic crises is high, and where suitable living-related donors are available.
Key Research Papers
- Strauss KA, Puffenberger EG, Morton DH. Maple Syrup Urine Disease. GeneReviews [Internet]. Seattle: University of Washington; 2006 (updated 2020) — Search PubMed
- Strauss KA, et al. Classical maple syrup urine disease and brain development: principles of management and formula design. Pediatrics. 2010;125(1):e229-38 — Search PubMed
- Barschak AG, et al. Oxidative stress in plasma from maple syrup urine disease patients during treatment. Mol Cell Biochem. 2008;309(1-2):169-77 — Search PubMed
- Feier FH, et al. Liver transplantation for maple syrup urine disease: a single institution experience. Liver Transpl. 2016;22(7):1032-41 — Search PubMed
- Mazariegos GV, et al. Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. Am J Transplant. 2012;12(5):1199-206 — Search PubMed
- Chuang DT, et al. Lessons from genetic disorders of branched-chain amino acid metabolism. J Nutr. 2006;136(1 Suppl):243S-249S. PMID: 16365098
- Morton DH, et al. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109(6):999-1008 — Search PubMed
- Bodner-Leidecker A, et al. Branched-chain L-amino acid metabolism in classical maple syrup urine disease after orthotopic liver transplantation. J Inherit Metab Dis. 2000;23(7):681-91 — Search PubMed
- Schonberger S, et al. Dependence of neurological outcome on early dietary management in maple syrup urine disease. J Inherit Metab Dis. 2004;27(4):441-53 — Search PubMed
- Parini R, et al. Liver transplant in maple syrup urine disease: a single-center experience. Liver Transpl. 2009;15(10):1173-9 — Search PubMed
- Snyderman SE. The therapy of maple syrup urine disease. Am J Dis Child. 1967;113(1):68-73 — Search PubMed
- Jouvet P, et al. Maple syrup urine disease metabolic crisis: clinical and biochemical analysis of a cohort of 20 classic neonatal forms. Eur J Pediatr. 2011;170(2):233-40 — Search PubMed
Search PubMed: Maple Syrup Urine Disease BCKAD
Connections
- Genetics
- Phenylketonuria
- Homocystinuria
- Gaucher Disease
- Amino Acids
- Lab Tests
- Neurology
- Gastroenterology