Pheochromocytoma: History and Discovery
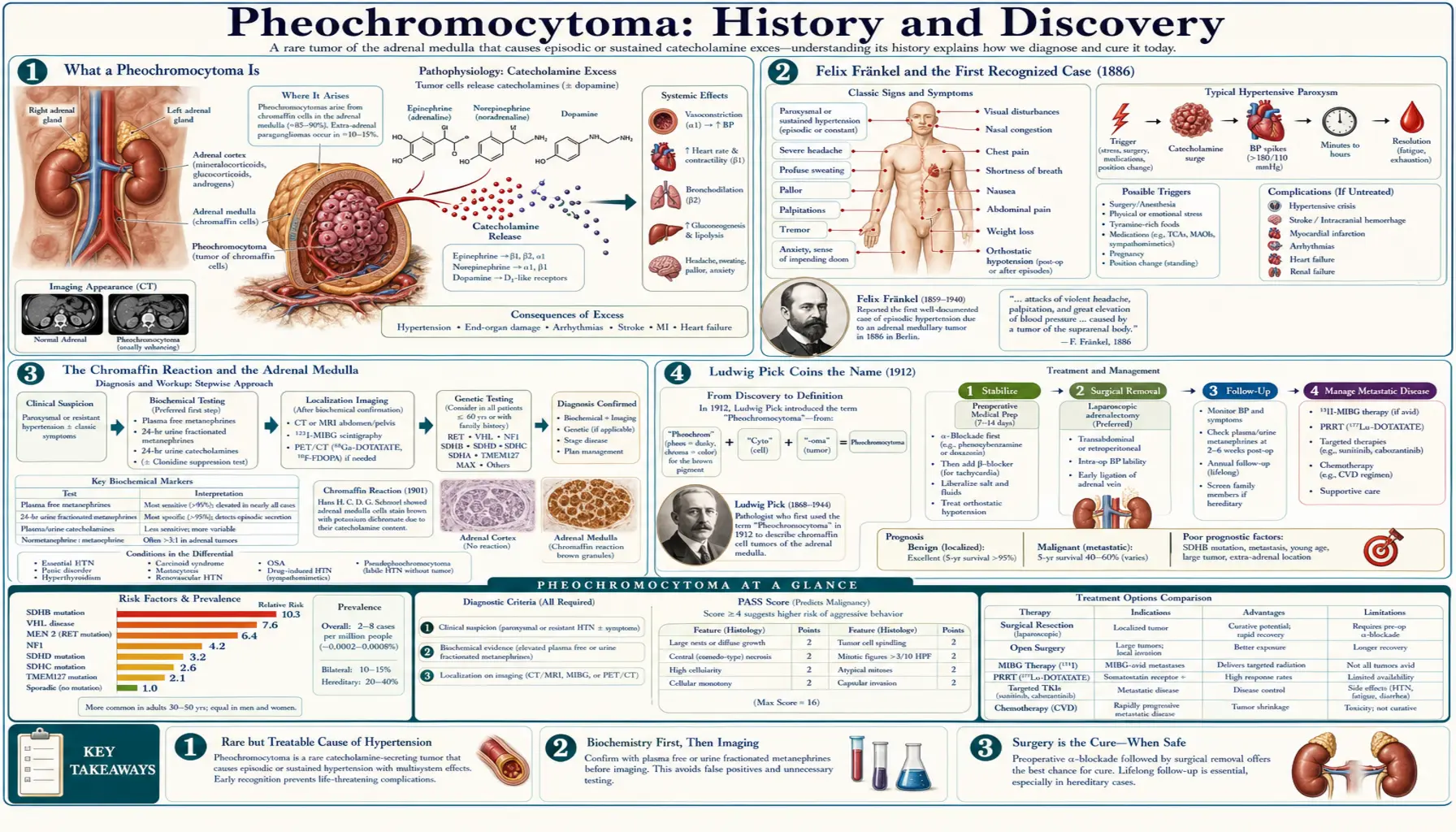
A pheochromocytoma is a usually benign, catecholamine-secreting tumour of the adrenal medulla — the inner core of the small glands that sit atop each kidney. By flooding the bloodstream with adrenaline (epinephrine) and noradrenaline, it produces dramatic episodic attacks of high blood pressure, pounding palpitations, drenching sweats, and severe headaches. Its story spans roughly a century and a half: the first recognized case was described by the German physician Felix Fränkel in 1886; the pathologist Ludwig Pick coined the name “pheochromocytoma” in 1912, from the Greek phaios (dusky) and chroma (colour), after the dark stain the tumour cells take with chromium salts; and the first patients were cured by surgery only in 1926. This page traces that history accurately, taking care to separate the first case from the naming, the chemistry, and the first operations.
Table of Contents
- What a Pheochromocytoma Is
- Felix Fränkel and the First Recognized Case (1886)
- The Chromaffin Reaction and the Adrenal Medulla
- Ludwig Pick Coins the Name (1912)
- Adrenaline: The Hormone Behind the Symptoms
- The First Successful Operations (1926)
- Building a Diagnosis Across the Twentieth Century
- The Rule of 10s and the Hereditary Syndromes
- Legacy: From Autopsy Curiosity to Curable Tumour
- Research Papers and References
- Connections
- Featured Videos
What a Pheochromocytoma Is
Each adrenal gland has two functionally distinct parts. The outer rind, the cortex, makes steroid hormones such as cortisol and aldosterone. The inner core, the medulla, is in effect a specialized piece of the sympathetic nervous system: its chromaffin cells manufacture and store the “fight-or-flight” catecholamines, chiefly adrenaline (epinephrine) and noradrenaline (norepinephrine). A pheochromocytoma is a tumour that arises from these chromaffin cells and, crucially, keeps making and releasing catecholamines outside the body’s normal control.
That single fact explains the whole clinical picture. When the tumour discharges a surge of catecholamines, blood vessels clamp down and the heart races, producing the classic paroxysm: a sudden spike of blood pressure, a hammering heartbeat, a pounding headache, profuse sweating, pallor, tremor, anxiety, and sometimes chest pain or a sense of impending doom. Between attacks a patient may feel entirely well, which is exactly what made the tumour so hard to recognize before anyone understood adrenal physiology. Most pheochromocytomas are benign and confined to one adrenal gland, but a minority are bilateral, arise outside the adrenal (where the same tumour is called a paraganglioma), or behave malignantly.
Understanding the disease therefore required three separate discoveries that did not arrive together: recognizing the tumour itself, recognizing what its cells were (chromaffin tissue), and recognizing the chemical messenger they released (adrenaline). The history below follows each thread in turn.
Felix Fränkel and the First Recognized Case (1886)
The first complete clinical-and-pathological description of what we now call a pheochromocytoma is credited to Felix Fränkel (rendered in older literature as Fraenkel), in a paper published in Virchows Archiv in 1886. His patient was an 18-year-old woman, Minna Roll, who was treated at the University Hospital of Freiburg and died there (the patient had died in 1884; Fränkel’s detailed report appeared in 1886). During life she had suffered exactly the paroxysms now considered classic — sudden palpitations, dizziness, headache, vomiting, weakness, and failing eyesight — and at autopsy she was found to have tumours of both adrenal glands. The paper’s original title described “a case of bilateral, completely latent adrenal tumour with concurrent nephritis,” capturing how silent the tumours had seemed until they killed her.
Two points deserve emphasis for accuracy. First, Fränkel did not coin the modern name and did not yet understand the catecholamine mechanism — adrenaline had not been isolated — yet, strikingly, he speculated that some substance normally present in the blood might be released in excess and “irritate” the vessels and organs, an intuition remarkably close to the truth. Second, the histological description in that work was largely the contribution of the Freiburg pathologist Max Schottelius, who examined the tumour microscopically and noted that it took on a brown colour in chromate-containing fixative — the very reaction that would later give the disease its name. Schottelius was not listed as an author and his role went under-recognized for over a century.
The 1886 case has a remarkable modern coda. In 2007 a New England Journal of Medicine report by Neumann and colleagues traced Minna Roll’s family and presented genetic evidence that her bilateral tumours reflected an inherited syndrome — multiple endocrine neoplasia type 2 (MEN-2), caused by a RET mutation. In other words, the very first described pheochromocytoma was, in hindsight, also one of the first documented hereditary ones — though that interpretation is a twenty-first-century reconstruction, not something Fränkel could have known.
The Chromaffin Reaction and the Adrenal Medulla
To understand why the tumour is named for a colour, one has to understand the staining quirk at the heart of the adrenal medulla. When tissue from the medulla — or from a pheochromocytoma — is treated with chromium salts (chromic acid or potassium dichromate, as in the historical Mueller’s fixative), the catecholamine-rich granules inside the cells are oxidized and turn a distinctive brown-to-dusky colour. This is the chromaffin reaction, and cells that show it are called chromaffin cells. The colour is essentially a chemical fingerprint of stored adrenaline and noradrenaline.
The term “chromaffin” itself was introduced by the Prague histologist Alfred Kohn (1867–1959) in 1902, in his work Das chromaffine Gewebe (“The chromaffin tissue”), and he went on, in 1903, to describe the related concept of paraganglia — collections of the same chromaffin tissue scattered outside the adrenal gland along the sympathetic chain. Kohn’s framework is why tumours of identical cells are today split by location into adrenal pheochromocytomas and extra-adrenal paragangliomas. His naming of the chromaffin reaction set the stage directly: once pathologists had a word for the dusky-staining cells, it was a short step to naming a tumour made of them.
Ludwig Pick Coins the Name (1912)
The name “pheochromocytoma” was coined by the Berlin pathologist Ludwig Pick in 1912. Pick built the word directly from the chromaffin reaction described above: from the Greek phaios (φαιος), meaning “dusky” or “dark,” plus chroma (χρῶμα), “colour,” plus cytoma, a tumour of cells — literally “the dusky-coloured-cell tumour.” The name memorializes the dark colour the tumour cells take on when exposed to chromium salts, exactly the feature Schottelius had observed under the microscope in 1886 and that Kohn had formalized in 1902.
It is worth being precise about who did what, because these milestones are often blurred together. Fränkel described the first case (1886); Kohn named the cell type, “chromaffin” (1902); and Pick named the tumour, “pheochromocytoma” (1912). None of these is the same event, and twenty-six years separated the first recognized case from the word now used for it the world over. The spelling “phaeochromocytoma” (with the classical ae diphthong) remains standard in British usage, while “pheochromocytoma” is the American form; both encode the same Greek roots.
Adrenaline: The Hormone Behind the Symptoms
A tumour that secretes a hormone cannot be fully understood until the hormone itself is known — and adrenaline was isolated only around the turn of the twentieth century, after Fränkel’s case but before Pick’s name. In the 1890s researchers had shown that extracts of the adrenal medulla powerfully raised blood pressure, pointing to an active principle. Around 1901, the Japanese-American chemist Jokichi Takamine (working with his assistant Keizo Uenaka) isolated a pure crystalline form of the active substance and marketed it, with Parke, Davis & Company, under the trade name Adrenalin; the American chemist Thomas Aldrich independently prepared the same crystalline compound and confirmed its identity that same year.
The naming history is genuinely tangled, and it is worth stating carefully. A few years earlier the American pharmacologist John Jacob Abel had worked on the adrenal principle and applied the name epinephrine, but the substance he isolated was later shown to be a benzoyl derivative rather than the pure hormone. This is the historical root of the enduring “adrenaline versus epinephrine” split — adrenaline (from Takamine’s trademark) became the common British name, while epinephrine (from Abel) became the official United States name for the identical molecule. By the time Pick named the tumour in 1912, then, the chemical it secreted had a name (in fact two), and physicians could begin to connect the dramatic blood-pressure attacks of Fränkel’s patient to a flood of this single, newly characterized hormone.
The First Successful Operations (1926)
Recognizing and naming the tumour was one thing; removing it safely was another, and far harder. Operating on a pheochromocytoma is uniquely dangerous because handling the tumour squeezes out catecholamines, sending blood pressure to lethal heights, and then removing it abruptly cuts off that hormonal support, causing a precipitous, sometimes fatal collapse. Before that physiology was understood or could be controlled, surgery was perilous.
The first successful surgical removals of a pheochromocytoma both occurred in 1926. The Swiss surgeon César Roux, chief of surgery at the cantonal hospital in Lausanne, is generally credited with the first, in February 1926. About seven months later, in October 1926, Charles H. Mayo of the Mayo Clinic performed the first successful removal in the United States. (Some accounts add the Yugoslav surgeon Isidor Papo among the earliest in the same period.) Several older and popular summaries place Mayo’s operation in 1927; the detailed surgical-history literature — including R. B. Welbourn’s authoritative 1987 review of the early surgical history — dates it firmly to October 1926, the same year as Roux and a few months later.
These pioneering operations succeeded partly by luck and partly by skill; the systematic medical preparation that makes the surgery routinely safe today — blocking the catecholamine receptors with alpha-adrenergic blockade for days before the operation, and managing blood pressure and volume meticulously throughout — was developed only in later decades. What the 1926 cases proved was the essential point: a pheochromocytoma, once found, could be cut out, and a patient otherwise destined to die could be cured.
Building a Diagnosis Across the Twentieth Century
For decades the tragedy of pheochromocytoma was that it was usually found at autopsy, not in the clinic — the “completely latent” tumour of Fränkel’s title. Turning it into a diagnosis the living could receive took the rest of the twentieth century and depended on biochemistry. Once adrenaline and noradrenaline were known, and especially once their breakdown products — the metanephrines and vanillylmandelic acid (VMA) — could be measured in blood and urine, physicians finally had a way to catch the tumour in the act of over-secreting. The pharmacological agent phentolamine was also used historically as a provocative/blocking test. Measurement of plasma free metanephrines later became the most sensitive screening test and is the modern standard.
Localization followed the broader arc of medical imaging. Where early surgeons had to explore the adrenal regions essentially blind or by crude means, the second half of the century brought CT and MRI to pinpoint an adrenal mass, and nuclear-medicine techniques — notably MIBG (metaiodobenzylguanidine) scanning, which is taken up specifically by chromaffin tissue, and later functional PET tracers — to find tumours that had spread or arisen outside the adrenal gland. The combination of a biochemical “is there too much catecholamine?” test with an anatomical “where is it?” scan is what finally made confident, pre-operative diagnosis the norm rather than the exception.
The Rule of 10s and the Hereditary Syndromes
As cases accumulated, clinicians distilled the tumour’s behaviour into a famous teaching mnemonic, the “rule of 10s”: roughly 10 percent of pheochromocytomas are bilateral, 10 percent are extra-adrenal (paragangliomas), 10 percent are malignant, 10 percent occur in children, and 10 percent are familial. The rule of 10s is a modern heuristic and a deliberate simplification — genuinely useful for learners, but it should be treated as an approximate memory aid rather than a precise statistic. Notably, modern genetics has overturned the “10 percent familial” figure in particular: it is now recognized that a much larger share of pheochromocytomas and paragangliomas — on the order of a third or more — carry an identifiable germline mutation, so the hereditary fraction is far higher than the old rule implied.
That genetic understanding is largely a development of the late twentieth and twenty-first centuries. Pheochromocytoma is now known to be a hallmark of several inherited cancer-predisposition syndromes, including multiple endocrine neoplasia types 2A and 2B (MEN-2), caused by RET mutations; von Hippel–Lindau (VHL) disease; neurofibromatosis type 1 (NF1); and the hereditary paraganglioma syndromes driven by mutations in the succinate-dehydrogenase (SDH) genes. The recognition that the very first case — Minna Roll’s bilateral tumours in 1886 — was almost certainly a MEN-2 case closes a striking historical loop: the disease that began as an unexplained autopsy finding is now, in many patients, traced to a single inherited gene.
Legacy: From Autopsy Curiosity to Curable Tumour
The history of pheochromocytoma is, at bottom, a history of three separate insights converging. In 1886 Felix Fränkel and Max Schottelius recognized the tumour; in 1902 Alfred Kohn named the cells it came from; around 1901 Takamine and others isolated the hormone those cells released; and in 1912 Ludwig Pick fused the chemistry and the pathology into a single evocative name — the “dusky-coloured” tumour. Only after all of that did surgery, in 1926, make it curable, and only across the following century did biochemistry, imaging, and genetics make it findable, treatable, and increasingly predictable.
For a patient today, that long history has a very practical payoff. A condition that once announced itself only through sudden, terrifying, unexplained attacks — and was often understood only after death — is now usually a diagnosable, operable, and frequently curable tumour, with hereditary forms that can be screened for in advance in at-risk families. The clinical details — symptoms, the metanephrine and imaging work-up, the crucial pre-operative alpha-blockade, surgery, and follow-up — are covered on the main Pheochromocytoma page. This history is offered as background and education and is not medical advice; anyone with episodic severe high blood pressure, pounding headaches, palpitations, and sweating should seek evaluation from a qualified clinician.
Research Papers and References
The references below combine landmark historical papers — including a translated reprint of Fränkel’s original 1886 description and the 2007 genetic re-examination of that family — with curated PubMed topic-search links into the history and biology of pheochromocytoma. Where a stable identifier is available it is given; otherwise the link opens a PubMed search at the National Library of Medicine in a new tab. Older primary works (Fränkel 1886; Kohn’s Das chromaffine Gewebe, 1902; Pick 1912) are named in the article as historical sources.
- Fränkel F. Classics in oncology. A case of bilateral completely latent adrenal tumor and concurrent nephritis with changes in the circulatory system and retinitis (1886; translated reprint). CA: A Cancer Journal for Clinicians. 1984;34(2):93-106. — PubMed: Fränkel 1886 classic case (reprint)
- Neumann HPH, Vortmeyer A, Schmidt D, et al. Evidence of MEN-2 in the original description of classic pheochromocytoma. New England Journal of Medicine. 2007;357(13):1311-1315. — doi:10.1056/NEJMoa071407
- Tischler AS, Pacak K, Eisenhofer G, et al. Max Schottelius: pioneer in pheochromocytoma. Journal of the Endocrine Society. 2017;1(7):957-964. — doi:10.1210/js.2017-00208
- Welbourn RB. Early surgical history of phaeochromocytoma. British Journal of Surgery. 1987;74(7):594-596. — doi:10.1002/bjs.1800740717
- Manger WM, Eisenhofer G. Pheochromocytoma: diagnosis and management update. Current Hypertension Reports — PubMed: Manger pheochromocytoma review
- Lenders JWM, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. The Lancet. 2005;366(9486):665-675. — doi:10.1016/S0140-6736(05)67139-5
- Parikh R, et al. ‘Great Masquerader’: a history of diagnosing pheochromocytoma. ANZ Journal of Surgery. 2025. — doi:10.1111/ans.19257
- Pheochromocytoma history and the naming by Ludwig Pick (1912); etymology phaios + chroma — PubMed: pheochromocytoma history and naming
- Alfred Kohn, the chromaffin reaction, and the chromaffin cell (1902) — PubMed: chromaffin cell history (Kohn)
- Abel, Takamine, and the isolation of adrenaline / epinephrine (c. 1901) — PubMed: isolation of adrenaline / epinephrine
- Hereditary pheochromocytoma and paraganglioma — MEN-2, VHL, NF1, and SDH genes — PubMed: hereditary pheochromocytoma and paraganglioma
- Plasma free metanephrines and the biochemical diagnosis of pheochromocytoma — PubMed: plasma metanephrines diagnosis
- Pre-operative alpha-adrenergic blockade and perioperative management of pheochromocytoma — PubMed: perioperative alpha-blockade
- MIBG scintigraphy and functional imaging of pheochromocytoma and paraganglioma — PubMed: MIBG and functional imaging
External Authoritative Resources
- National Cancer Institute — Pheochromocytoma and Paraganglioma
- NIH GARD — Pheochromocytoma
- PubMed — All research on the history of pheochromocytoma
Connections
- Endocrinology
- Pheochromocytoma (main page)
- Hypertension
- Addison’s Disease
- Cushing’s Syndrome
- Diabetes
- All Conditions