Brain Aging: History and Origins
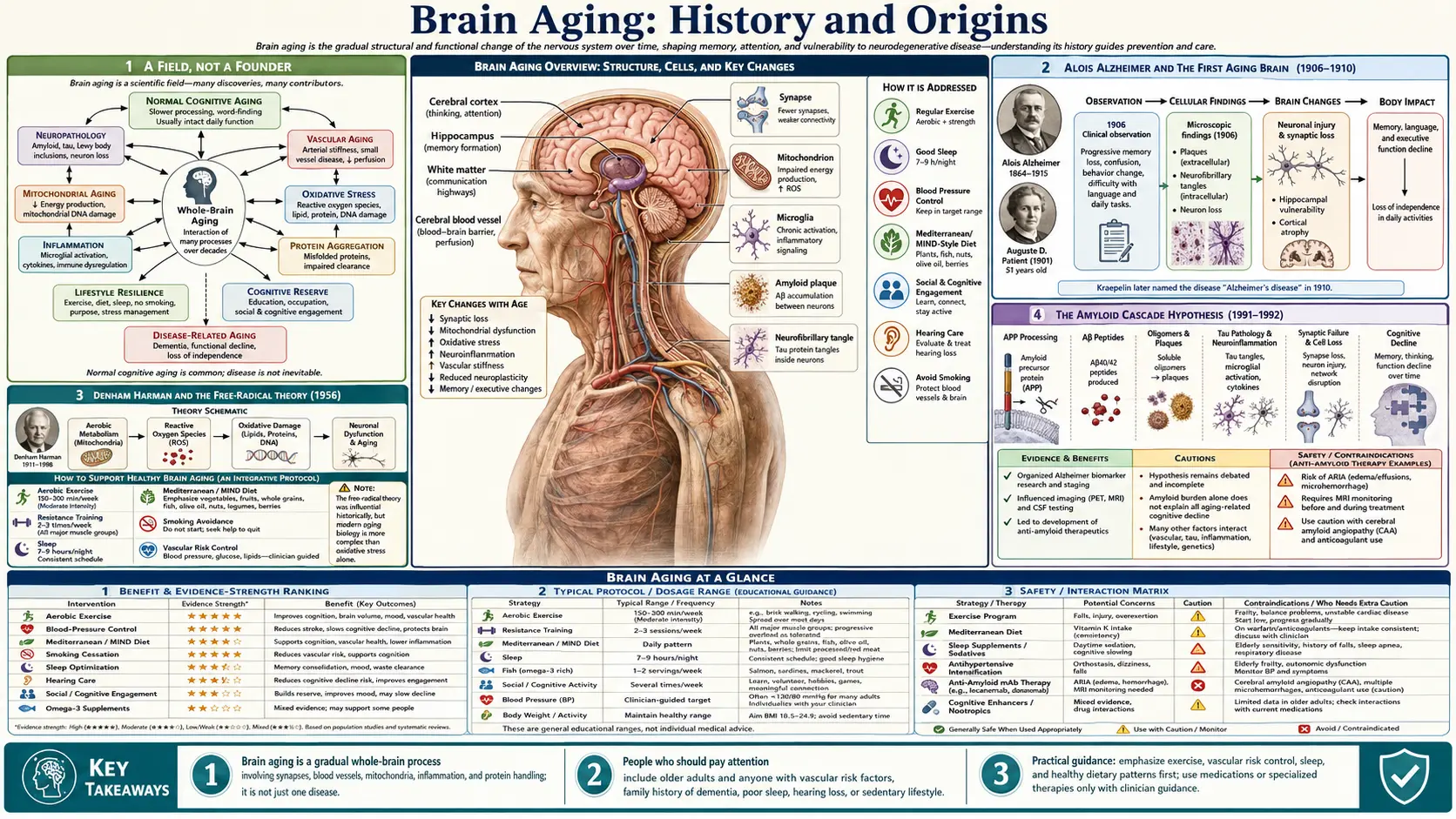
Unlike a herb or a branded protocol, "brain aging" has no single inventor and no founding date. It is a field of science, and its story is the slow, century-long effort to answer one question: what actually happens inside an aging brain, and how much of it can be slowed? This history traces that effort honestly — from Alois Alzheimer's first autopsy in 1906 and Denham Harman's free-radical theory of 1956, through the amyloid hypothesis of the 1990s, the discovery of the brain's waste-clearing "glymphatic" system in 2012, the epigenetic clocks that finally let us measure biological age, and the modern realization that a large fraction of dementia is preventable. Where a date, a name, or a claim is firmly documented we say so; where the science is still argued over — and much of it is — we say that too. This is a story of accumulating evidence, not of a cure.
Table of Contents
- A Field, Not a Founder
- Alois Alzheimer and the First Aging Brain (1906–1910)
- Denham Harman and the Free-Radical Theory (1956)
- The Amyloid Cascade Hypothesis (1991–1992)
- The Glymphatic System: A Brain That Cleans Itself (2012–2013)
- The Hallmarks of Aging Reach the Brain (2013–2023)
- Measuring Biological Age: The Epigenetic Clocks (2013–)
- From Inevitable to Modifiable: The Prevention Era (2015–2024)
- Evidence and Honest Caveats
- Research Papers and References
- Connections
- Featured Videos
A Field, Not a Founder
It is worth being clear at the outset about what kind of history this is. Many of the topics on this site have a real, named founder — a physician who built a therapy, an inventor who isolated a molecule. Brain aging is not one of them. No one person discovered it, named it, or owns it. It is a scientific subject that thousands of researchers have built up, piece by piece, over more than a hundred years, and anyone who claims to have a single "protocol" that solves it is overstating what the science supports.
What a history of brain aging can honestly trace is the sequence of discoveries that changed how the question was understood. For most of human history, the mental decline of old age was simply assumed to be an inevitable, untreatable part of growing old — "senility," a single fate awaiting everyone who lived long enough. The intellectual story of the last century is the gradual dismantling of that assumption: the recognition that the aging brain undergoes specific, measurable molecular changes; that some of those changes drive distinct diseases while others reflect ordinary wear; and — most recently and most importantly — that a meaningful share of late-life cognitive decline is not fixed at all, but can be delayed.
The sections that follow are organized around the people and papers that marked the turning points. They are presented as milestones in an unfinished investigation, not as settled truths. The detailed mechanisms, biomarkers, and intervention evidence are covered on the main Brain Aging page; this article is concerned with how each of those ideas first arrived.
Alois Alzheimer and the First Aging Brain (1906–1910)
The modern study of the aging brain has a reasonable starting point: the work of the German psychiatrist and neuropathologist Alois Alzheimer (1864–1915). In 1901, while working in Frankfurt, Alzheimer admitted a 51-year-old patient named Auguste Deter, who suffered from profound memory loss, language difficulty, disorientation, and paranoia. He followed her case closely until her death in 1906. When he examined her brain after death, he found two features that would become the defining signatures of the disease later named for him: shrinkage of the cortex, dense deposits between the nerve cells (now called amyloid plaques), and tangled fibres inside them (now called neurofibrillary tangles).
Alzheimer presented these findings publicly on 3 November 1906 at a meeting of southwest German psychiatrists in Tübingen, and published a fuller account in 1907. The condition acquired its name from his influential supervisor, the psychiatrist Emil Kraepelin, who included it as "Alzheimer's disease" in the 1910 eighth edition of his standard textbook of psychiatry. Historians have noted, in the interest of accuracy, that Kraepelin may have had institutional and funding motives for highlighting the case as a strikingly novel disease, and that a contemporary, the Prague pathologist Oskar Fischer, was independently describing very similar plaque pathology at almost the same time — a reminder that scientific "firsts" are often shared.
Two things make this the right place to begin a history of brain aging, even though Alzheimer studied a disease rather than ordinary aging. First, he established that the decline of the aging brain has a physical, microscopic substrate — something you can see under a lens — rather than being a vague failure of vitality. Second, the very plaques and tangles he drew in 1906 are the same proteins (beta-amyloid and tau) that sit at the center of brain-aging research more than a century later. The thread from his hand-drawn slides to today's blood tests for these proteins is unbroken.
Denham Harman and the Free-Radical Theory (1956)
If Alzheimer connected brain decline to visible pathology, the next great step was to ask what causes aging at the chemical level — a question that applies to the whole body, the brain included. The most influential answer came from an American chemist and physician, Denham Harman (1916–2014), who in 1956 published a short, now-famous paper in the Journal of Gerontology titled "Aging: a theory based on free radical and radiation chemistry."
Harman's proposal was elegant and, for its time, radical. He suggested that aging is driven by cumulative damage from free radicals — highly reactive molecules generated as an unavoidable by-product of using oxygen to make energy. Over a lifetime, he argued, these reactive species chip away at the body's cells and tissues, and that accumulated oxidative damage is a root cause of aging and the diseases that come with it. Harman had trained as a chemist before becoming a physician, and he drew the idea from radiation chemistry, where free radicals were already understood to damage biological molecules.
The theory is directly relevant to the brain for a simple reason: the brain consumes a disproportionate share of the body's oxygen, which makes it an unusually rich source of the very free radicals Harman described. His framework gave rise to the entire concept of oxidative stress in the nervous system and underwrote decades of interest in antioxidants. It is important to be honest about how the idea has aged, however. The simple version — "take antioxidants, slow aging" — has largely failed in large human trials, and the modern view is more nuanced: some free-radical signaling is necessary and even beneficial, and indiscriminate antioxidant supplementation has not extended human lifespan. Harman's theory remains a foundational landmark in the history of aging science, but it is now treated as one important contributor to a far more complicated picture, not the whole answer.
The Amyloid Cascade Hypothesis (1991–1992)
For most of the twentieth century, the plaques Alzheimer had drawn were known but not explained. That changed at the end of the 1980s and start of the 1990s, when molecular biology made it possible to identify the proteins involved and to study families in which Alzheimer's disease was inherited. Out of this work came the single most influential — and most fiercely debated — idea in the field: the amyloid cascade hypothesis.
The hypothesis holds that the accumulation of the protein fragment beta-amyloid is the initiating event in Alzheimer's disease, setting off a downstream cascade that includes tau tangles, inflammation, and the death of neurons. The idea took shape across several papers, and honesty about its authorship matters: the neuroscientist Dennis Selkoe laid out much of the molecular groundwork in 1991, and the geneticists John Hardy and Gerald Higgins gave it its enduring name in a concise 1992 paper in Science titled "Alzheimer's disease: the amyloid cascade hypothesis." The strongest early evidence came from rare inherited forms of the disease, in which mutations affecting amyloid production cause aggressive, early-onset Alzheimer's.
The amyloid hypothesis dominated research and drug development for thirty years, and its history is genuinely contested. Dozens of drugs designed to remove amyloid failed in trials, leading many scientists to question whether amyloid was truly the cause or merely a tombstone marking damage done by something else. The debate is not over. What shifted the picture recently was the arrival, in the 2020s, of antibody drugs (lecanemab and donanemab) that clear amyloid and produce a real but modest slowing of decline — on the order of a quarter to a third over eighteen months in early disease. That outcome is widely read as partial vindication: amyloid does appear to matter, but it is clearly not the entire story, and tau pathology tracks cognitive decline more closely. The careful conclusion, echoed in recent reviews, is that the amyloid cascade remains a working hypothesis — influential and partly supported, but not proven to be the sole driver of the disease.
The Glymphatic System: A Brain That Cleans Itself (2012–2013)
One of the most striking recent additions to the history of brain aging arrived in 2012, when a team led by the Danish neuroscientist Maiken Nedergaard at the University of Rochester described a previously unrecognized waste-clearance network in the brain. The brain has no conventional lymphatic vessels, yet it is the body's most metabolically active organ and must somehow dispose of its waste, including the very amyloid and tau proteins implicated in dementia. Nedergaard's group showed that cerebrospinal fluid flows along the spaces around blood vessels, washes through the brain tissue with the help of channels (aquaporin-4) on supportive cells called astrocytes, and carries solutes away.
They named it the glymphatic system — a blend of "glia" (the brain's non-neuronal cells) and "lymphatic." By documented accounts, the term was coined by Nedergaard's collaborator and husband, the neurologist Steven Goldman. The founding paper, by Jeffrey Iliff, Nedergaard, and colleagues, appeared in Science Translational Medicine in August 2012. A year later, in a 2013 paper in Science, the group reported an even more arresting finding: this cleaning happens mostly during sleep. During deep sleep the spaces between brain cells widen, fluid exchange roughly doubles, and the clearance of injected amyloid accelerates.
The relevance to brain aging is immediate. Glymphatic flow appears to decline with age and to be impaired in Alzheimer's disease, and the discovery gave a concrete biological reason why chronic poor sleep and untreated sleep apnea are linked to cognitive decline — if the brain cleans itself during sleep, sleep loss may let waste accumulate. It is fair to add a note of scientific caution: glymphatic research is young, some of its mechanics are still being refined and debated, and most of the strongest data come from animals. But the discovery genuinely reshaped how the field thinks about the aging brain, turning sleep from a lifestyle suggestion into a candidate mechanism.
The Hallmarks of Aging Reach the Brain (2013–2023)
By the early 2010s, aging research had grown into a sprawling collection of separate findings — on DNA damage, telomeres, mitochondria, senescent cells, and more — that badly needed an organizing framework. That framework arrived in 2013, in a landmark review titled "The Hallmarks of Aging," published in Cell by the Spanish molecular biologist Carlos López-Otín and his colleagues Maria Blasco, Linda Partridge, Manuel Serrano, and Guido Kroemer. The paper proposed that aging in every organ could be understood through nine shared hallmarks: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem-cell exhaustion, and altered intercellular communication.
The hallmarks paper became one of the most cited works in all of biology, precisely because it gave researchers a common language. For brain aging, its value was to show that the brain is not exempt from the general rules of aging — it simply expresses them through neuron-specific failures. The protein-aggregation hallmark (loss of proteostasis) maps onto the plaques and tangles of Alzheimer's; mitochondrial dysfunction is central to Parkinson's; cellular senescence and inflammation drive the "inflammaging" of the aged brain. In 2023, López-Otín and the same colleagues published an update, "Hallmarks of Aging: An Expanding Universe," expanding the list to twelve by adding disabled autophagy, chronic inflammation, and microbiome disturbance (dysbiosis) — the last reflecting the growing recognition of the gut–brain connection in neuroinflammation.
This framework is the bridge between general longevity science and brain-specific prevention. It explains why the same interventions studied for whole-body aging — exercise, caloric restriction, drugs like rapamycin that target nutrient-sensing — keep reappearing in discussions of cognitive aging. It also keeps the field honest: the hallmarks are a map of damage mechanisms, and a map is not a treatment. Identifying twelve hallmarks does not mean we can fix any of them on demand.
Measuring Biological Age: The Epigenetic Clocks (2013–)
A recurring problem in aging research was the absence of a good ruler. Chronological age — how many birthdays you have had — is a poor measure of how aged your tissues actually are, and earlier biological markers such as telomere length proved too noisy to be useful. The breakthrough came in 2013, the same remarkable year as the hallmarks paper and the glymphatic-sleep study, when the geneticist Steve Horvath at UCLA published an "epigenetic clock" in the journal Genome Biology.
Horvath's insight was that chemical tags called DNA methylation accumulate on the genome in remarkably predictable patterns as we age. By measuring methylation at a few hundred specific sites, his clock could estimate a tissue's age with a median error of under four years — and, crucially, it worked across nearly all human tissues, including the brain. For the first time there was a quantitative measure of biological aging. Horvath later developed a brain-specific version trained on cortex tissue, which yields a "brain age gap" — the difference between a brain's methylation age and the person's actual age — that independently predicts cognitive decline.
The clocks set off a wave of development. Second-generation versions trained to predict health and death rather than just age — PhenoAge (Morgan Levine and colleagues, 2018), GrimAge (Ake Lu, Horvath, and colleagues, 2019), and DunedinPACE (Daniel Belsky and colleagues, 2022, which measures the current pace of aging) — predict mortality and dementia risk better than the original. The honest caveat, repeated by the clocks' own developers, is essential here: these tools are biomarkers, not mechanisms. A clock that says a brain is aging fast tells you something is wrong; it does not tell you what, and merely "turning back the clock" on a readout is not the same as repairing the underlying damage. The clocks made aging measurable — an enormous advance — but they did not make it curable.
From Inevitable to Modifiable: The Prevention Era (2015–2024)
Perhaps the most consequential shift in the modern history of brain aging is not a molecule or a mechanism but a change of expectations — the growing evidence that a large share of dementia is preventable. Two strands of work drove this change. The first was direct clinical proof. In 2015, a Finnish team led by Tiia Ngandu and Miia Kivipelto published the FINGER trial in The Lancet — the first large randomized controlled trial to show that a combined program of diet, exercise, cognitive training, and management of vascular risk could measurably protect cognition in at-risk older adults. FINGER turned a hopeful idea into demonstrated fact and spawned a worldwide network of replication studies.
The second strand was the work of synthesis. Beginning in 2017, the Lancet Commission on dementia prevention, intervention, and care, led by the psychiatrist Gill Livingston, gathered the evidence on modifiable risk factors into a single influential accounting. Its first report identified nine factors responsible for roughly 35% of dementia cases. The 2020 update expanded the list to twelve factors (about 40%), adding excessive alcohol, traumatic brain injury, and air pollution. The 2024 update reached fourteen factors and up to 45% of cases, newly including untreated vision loss and high cholesterol. The full modern list spans the whole lifespan: education, hearing loss, high cholesterol, depression, head injury, physical inactivity, diabetes, smoking, hypertension, obesity, excess alcohol, air pollution, social isolation, and vision loss.
This is the most hopeful chapter in the story, and it deserves to be stated plainly because it is also the best-supported. For the first time in the history of the subject, brain aging is no longer treated as a uniform, untreatable fate. A great deal of late-life decline traces to factors a person can act on — blood pressure, blood sugar, hearing, sleep, exercise, social connection — and the highest-impact of these are also the cheapest and most accessible. None of it is a guarantee, and none of it reverses established dementia. But the journey from Alzheimer's 1906 verdict of inevitable senility to the 2024 finding that nearly half of dementia may be preventable is the genuine arc of this history.
Evidence and Honest Caveats
Because "brain aging" attracts an enormous amount of supplement marketing and longevity hype, an honest history has to end by separating what the evidence supports from what it does not. The discoveries above are real and well documented. What is far less certain is the leap from mechanism to intervention — and that leap is where most overclaiming happens.
What is genuinely well-supported. The prevention findings are the most solid part of the field: treating hypertension, diabetes, hearing loss, and depression; getting regular aerobic exercise; not smoking; staying socially and cognitively engaged; and sleeping adequately are all backed by strong epidemiology and, for the multidomain package, by randomized trials such as FINGER. The diet evidence (Mediterranean and MIND patterns) is good though largely observational. These are the interventions with real human data behind them.
What is promising but unproven. Most of the headline-grabbing compounds — NAD+ precursors, senolytics, rapamycin for longevity, urolithin A, spermidine, lithium microdoses — rest mainly on animal studies and small or short human trials. They are legitimate research directions, not established treatments, and rapamycin and senolytics in particular are investigational and not approved for slowing brain aging. The GLP-1 drugs and anti-amyloid antibodies have real trial data but for specific, narrow indications; their broader role in prevention is still being tested, with key Alzheimer's-prevention results expected mid-decade.
What to be skeptical of. Any product or program marketed as a proven way to "reverse brain aging," restore youthful cognition, or cure dementia is running ahead of the evidence. The epigenetic clocks are measurement tools, not proof that any supplement which nudges them is rejuvenating the brain. The free-radical theory's commercial offspring — the antioxidant-supplement boom — is a cautionary tale: a sound mechanism (oxidative damage) produced a popular intervention (antioxidant pills) that largely failed when tested rigorously. The lesson that runs through this entire history is the one worth keeping: a compelling mechanism is a reason to investigate, never a substitute for the trial that tests whether it actually helps people. Anyone making decisions about their own cognitive health should weigh this evidence with a clinician rather than a sales page.
Research Papers and References
The list below gathers the primary papers behind the milestones described above, followed by curated PubMed topic-search links and authoritative resources. Author names, titles, and journals are given as plain text; only the stable DOI or PubMed identifier is hyperlinked, and each opens in a new tab. The early historical sources — Alzheimer's 1906 presentation and 1907 paper, and Kraepelin's 1910 textbook — are named in the article as historical documents rather than as modern citations.
- Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298-300. — doi:10.1093/geronj/11.3.298 · PMID: 13332224
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184-185. — doi:10.1126/science.1566067 · PMID: 1566067
- Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Science Translational Medicine. 2012;4(147):147ra111. — doi:10.1126/scitranslmed.3003748 · PMID: 22896675
- Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373-377. — doi:10.1126/science.1241224 · PMID: 24136970
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-1217. — doi:10.1016/j.cell.2013.05.039 Search PubMed
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243-278. — doi:10.1016/j.cell.2022.11.001 Search PubMed
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biology. 2013;14(10):R115. — doi:10.1186/gb-2013-14-10-r115 · PMID: 24138928
- Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11(2):303-327. — doi:10.18632/aging.101684 · PMID: 30669119
- Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. The Lancet. 2015;385(9984):2255-2263. — doi:10.1016/S0140-6736(15)60461-5 · PMID: 25771249
- Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. The Lancet. 2017;390(10113):2673-2734. — doi:10.1016/S0140-6736(17)31363-6 Search PubMed
- Livingston G, Huntley J, Liu KY, et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. The Lancet. 2024;404(10452):572-628. — doi:10.1016/S0140-6736(24)01296-0 · PMID: 39096926
- History and ethics of Alzheimer's disease — Auguste Deter, Alois Alzheimer, and Kraepelin PubMed: Alois Alzheimer and Auguste Deter history
- Glymphatic system, sleep, and brain aging PubMed: glymphatic system, sleep, and brain aging
External Authoritative Resources
- National Institute on Aging (NIA) — Alzheimer's Disease Fact Sheet
- NCCIH — Cognitive Function, Dementia, and Alzheimer's Disease
- PubMed — Brain aging mechanisms and hallmarks
Connections
- All Remedies
- Brain Aging
- Longevity Protocols
- Rapamycin
- NAD+ / NMN
- Gut–Brain Axis
- Alzheimer's Disease
- Parkinson's Disease
- Homocysteine