Giant Cell Arteritis
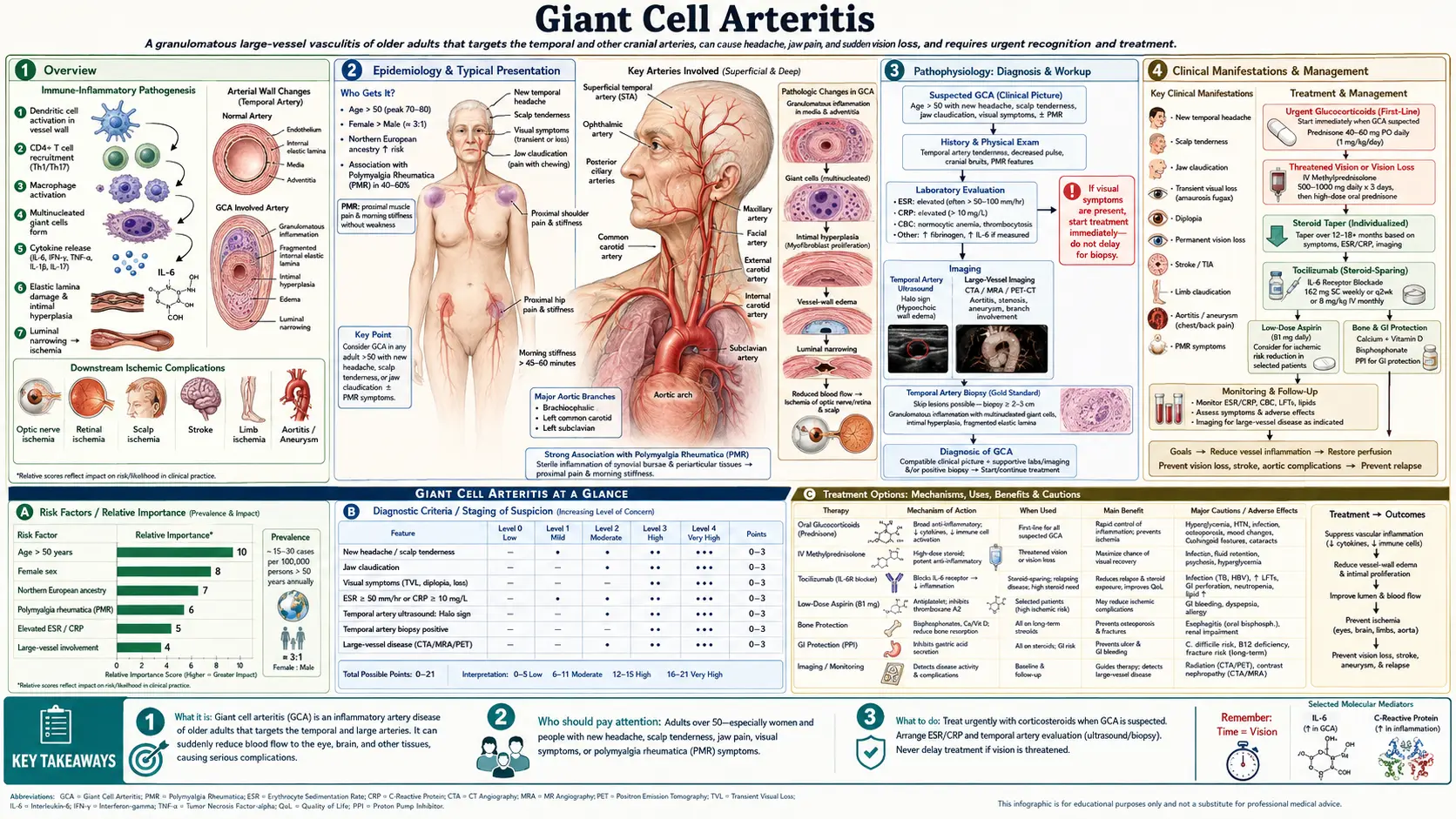
- Overview
- Epidemiology
- Pathophysiology
- Clinical Manifestations
- Diagnosis
- ACR 2022 Classification Criteria
- Treatment
- GCA–PMR Overlap
- Prognosis and Monitoring
- References
- Connections
Overview
Giant cell arteritis (GCA) is the most common primary systemic vasculitis in adults over the age of 50 in developed countries. It is a granulomatous inflammatory disease affecting large and medium-sized vessels, with a particular predilection for branches of the external and internal carotid arteries, the aorta, and its major branches. The condition was formerly known as "temporal arteritis" because of its frequent involvement of the temporal arteries, but this older name has largely been replaced as the full scope of large-vessel involvement became recognized. GCA affects women three times more often than men, with an overall incidence of 15 to 30 per 100,000 individuals over age 50, and occurs almost exclusively in people of Caucasian descent, particularly those of Northern European ancestry.
Cranial GCA — involving the temporal, ophthalmic, posterior ciliary, and vertebral arteries — is the most clinically urgent form of the disease. Inflammation and occlusion of the posterior ciliary arteries causes anterior ischemic optic neuropathy (AION), which is the leading cause of sudden, permanent vision loss in adults over 50. Because visual loss, once established, is almost never reversible, GCA must be treated as a true ophthalmologic emergency whenever visual symptoms are present. Prompt recognition and immediate initiation of high-dose corticosteroids before further ischemic events occur is the most critical intervention in clinical management.
Large-vessel GCA represents a second major disease phenotype in which granulomatous inflammation extends to the aorta, subclavian, axillary, and other proximal arteries. Patients with large-vessel involvement may present with arm claudication, blood pressure asymmetry between limbs, bruits over affected vessels, and, as a late complication, thoracic aortic aneurysm. Large-vessel GCA is often under-recognized clinically and may be identified incidentally on imaging studies performed for other indications. Some patients have overlapping cranial and large-vessel involvement, while others present exclusively with one phenotype.
Epidemiology
GCA occurs almost exclusively in individuals over age 50, with the mean age at onset typically ranging from 72 to 76 years. The disease is extraordinarily rare below age 50. Incidence rises sharply with advancing age, and the highest population-based rates have been documented in Olmsted County, Minnesota — a predominantly Scandinavian-descent cohort — where rates approach 29 per 100,000 individuals over age 50. Rates are substantially lower in Mediterranean European populations and are rare in African American, Asian, and Hispanic populations. This striking geographic and ethnic gradient strongly implicates both genetic and environmental determinants.
A genetic predisposition is well established. The HLA-DRB1*04 allele — particularly the shared epitope characteristic of rheumatoid arthritis susceptibility — is significantly associated with GCA and may influence disease expression and severity. Non-MHC genetic loci in immune regulatory pathways have also been identified in genome-wide association studies. Environmental triggers, possibly including certain infectious agents, have been proposed based on reports of seasonal clustering and geographic cycles in incidence, though no specific pathogen has been confirmed.
The lifetime risk of developing GCA is approximately 1% in women and 0.5% in men, reflecting the marked female predominance observed across all populations studied. Concurrent polymyalgia rheumatica (PMR) occurs in 40 to 50% of GCA patients at some point during their illness. Conversely, approximately 15 to 20% of patients diagnosed with pure PMR will eventually develop clinical features of GCA, and subclinical large-vessel vasculitis detectable on imaging may be present in a higher proportion of PMR patients. The two conditions share pathophysiologic mechanisms and likely represent a disease spectrum rather than entirely distinct entities.
Pathophysiology
The initiating event in GCA is thought to occur at the adventitial vasa vasorum — the small blood vessels that supply the walls of large arteries. Mature dendritic cells residing in the adventitia become inappropriately activated, possibly in response to an environmental antigen, and recruit CD4-positive T lymphocytes into the vessel wall. These T cells differentiate along both Th1 and Th17 pathways. Th1 cells produce interferon-gamma (IFN-γ), which drives macrophage activation and the formation of epithelioid granulomata containing multinucleated giant cells — the histological hallmark of the disease. Th17 cells, producing IL-17, contribute to a distinct inflammatory axis that may be relatively more steroid-resistant. Interleukin-6, produced in large quantities by activated macrophages and stromal cells, drives the systemic inflammatory response and accounts for the pronounced elevation of acute-phase reactants (ESR, CRP) characteristic of active GCA.
Activated macrophages in the media and intima of affected vessels produce platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), which stimulate smooth muscle cell proliferation and neoangiogenesis within the vessel wall. This process leads to progressive intimal hyperplasia and luminal stenosis, which is the final common pathway for the ischemic complications of GCA — including visual loss, jaw claudication, scalp necrosis, and stroke. The fragmentation of the internal elastic lamina, prominently visible on histology, is a direct consequence of matrix metalloproteinase release by activated macrophages and contributes to the structural disruption of the arterial wall.
The anatomical distribution of GCA reflects the biology of the vasa vasorum. Arteries with rich adventitial vasa vasorum — particularly the superficial temporal, posterior ciliary, ophthalmic, vertebral, subclavian, and aortic wall — are most vulnerable to inflammatory infiltration. Conversely, capillaries, venules, and vessels lacking a vasa vasorum are spared. This explains why GCA does not cause glomerulonephritis or cutaneous vasculitis despite being a systemic inflammatory disease. The selectivity of vascular involvement has important implications for understanding disease complications and targeting therapy.
Clinical Manifestations
The hallmark presentation of cranial GCA combines new-onset headache with one or more features of cranial ischemia or scalp and artery tenderness. The headache is typically described as temporal, often throbbing or boring in character, and may be severe and unremitting. Scalp tenderness — particularly tenderness over the course of the temporal artery — is frequently reported; patients may note discomfort when combing their hair or wearing a hat. On physical examination, the temporal artery may be thickened, nodular, pulseless, or exquisitely tender. These findings, when present, are highly suggestive of the diagnosis, though their absence does not exclude it.
Jaw claudication — pain or fatigue of the jaw muscles (masseter and temporalis) that comes on during chewing and is relieved by rest — is one of the most specific symptoms of GCA, occurring in approximately 40 to 50% of patients. It results from ischemia of the muscles of mastication supplied by branches of the external carotid artery. Tongue claudication and difficulty swallowing are less common but follow the same ischemic mechanism. Visual symptoms represent the most feared manifestation of GCA. Transient monocular visual loss (amaurosis fugax) may herald impending permanent visual loss. Established visual loss due to anterior ischemic optic neuropathy — caused by occlusion of the posterior ciliary arteries supplying the optic nerve head — typically presents as sudden, painless, monocular vision loss, often described as a curtain falling or a portion of the visual field going dark. Diplopia due to ischemia of extraocular muscles or cranial nerve VI palsy also occurs.
Constitutional symptoms are present in the majority of patients and may dominate the clinical picture before cranial symptoms emerge. Fatigue, malaise, unintentional weight loss, low-grade fever, and night sweats are common. An elevated erythrocyte sedimentation rate in an elderly patient with new headache and constitutional symptoms should always prompt consideration of GCA. Polymyalgia rheumatica — aching and morning stiffness of the shoulder and hip girdles — coexists in 40 to 50% of GCA patients and may be the presenting complaint.
Large-vessel GCA presents with symptoms referable to the subclavian, axillary, and brachial arteries: arm claudication with exertion, asymmetric blood pressure between the two arms (a difference exceeding 10 mmHg is clinically significant), subclavian or axillary bruits on auscultation, and reduced or absent radial pulses. Aortic involvement may be clinically silent initially but carries a 2- to 3-fold increased lifetime risk of thoracic aortic aneurysm compared with the general population — a late complication requiring long-term cardiovascular surveillance. Some patients present primarily with systemic inflammation and fever of unknown origin without cranial features, underscoring the protean nature of this disease.
Diagnosis
Laboratory evaluation in active GCA typically reveals a markedly elevated erythrocyte sedimentation rate — most commonly greater than 50 mm/hr and frequently exceeding 100 mm/hr — combined with elevated C-reactive protein. A normal ESR does not exclude GCA; approximately 5 to 10% of biopsy-proven cases present with a normal ESR, and CRP may be more sensitive in this context. Additional findings include normochromic normocytic anemia of chronic inflammation, reactive thrombocytosis, elevated alkaline phosphatase, and hypoalbuminemia. These are markers of systemic inflammation and are not specific to GCA.
Temporal artery biopsy remains the gold standard for diagnosis. A segment of at least 1.5 to 3 cm of the temporal artery should be excised to maximize the yield; shorter segments increase the risk of missing skip lesions. Histological findings of GCA include transmural inflammation with lymphocytes, macrophages, and multinucleated giant cells, fragmentation or disruption of the internal elastic lamina, and intimal hyperplasia with luminal narrowing. Skip lesions — segments of normal artery interspersed with involved segments — occur in approximately 5% of cases, which is the rationale for considering contralateral biopsy when the first biopsy is negative despite strong clinical suspicion. A critical practical point: corticosteroids do not immediately eliminate histological changes, and biopsy remains diagnostically positive for at least 2 weeks after initiation of treatment. Steroids must never be delayed awaiting biopsy results in a patient with threatened vision.
Imaging has transformed the diagnostic workup of GCA, particularly in centers with expertise in vascular ultrasound. Duplex ultrasonography of the temporal and axillary arteries demonstrating the "halo sign" — a concentric, hypoechoic ring of wall thickening surrounding the artery lumen, visible in longitudinal and transverse planes and not compressible with probe pressure — is now recognized by ACR/EULAR guidelines as an alternative to biopsy in experienced hands. FDG-PET/CT is the preferred imaging modality for detecting large-vessel GCA, demonstrating increased metabolic activity along the aortic wall, subclavian, and axillary arteries. MRI and MRA can also characterize vessel wall inflammation and luminal changes. Color Doppler ultrasound, PET, and MRI are complementary; choice of modality depends on the clinical phenotype (cranial vs. large-vessel) and local expertise.
ACR 2022 Classification Criteria
The American College of Rheumatology and EULAR jointly published updated classification criteria for GCA in 2022. These criteria are intended for classifying patients for research purposes and should not be used as standalone diagnostic criteria — a distinction that is clinically important. Classification requires that the patient be age 50 or older with clinical evidence of vasculitis, after other causes have been reasonably excluded.
The criteria use a point-based scoring system:
- Cranial symptoms: New onset of headache, scalp tenderness, or temporal artery tenderness, thickening, or nodularity — +2 points
- Jaw or tongue claudication: Pain or fatigue of jaw or tongue muscles during use — +2 points
- Visual loss attributable to GCA: Sudden, monocular visual loss consistent with ischemic optic neuropathy or retinal artery occlusion — +3 points
- Elevated acute-phase reactants: ESR ≥50 mm/hr or CRP ≥10 mg/L — +3 points
- Positive temporal artery biopsy: Histological evidence of granulomatous arteritis — +5 points
- Temporal artery ultrasound halo sign: Bilateral halo sign or halo sign plus compression sign in at least one temporal artery — +5 points
- PET evidence of large-vessel inflammation: FDG uptake in aorta or its branch vessels above background — +3 points
- PMR features: Bilateral shoulder and/or hip girdle aching with morning stiffness — +2 points
A total score of 6 or more points classifies the patient as having GCA. It is important to remember that these classification criteria were developed to ensure homogeneous patient populations in clinical trials, not to replace clinical judgment in individual patient management.
Treatment
The cornerstone of GCA treatment is immediate initiation of high-dose corticosteroids, and this must not be delayed under any circumstances when visual symptoms are present or when the diagnosis is strongly suspected. For cranial GCA with visual threat — including any history of transient visual loss, established visual loss, or jaw claudication suggesting critical ischemia — the recommended starting dose is prednisone 60 mg per day (or 1 mg/kg/day, up to 60 mg). For patients presenting with acute visual loss or bilateral visual involvement, intravenous methylprednisolone 1 g per day for three consecutive days should be administered before transitioning to oral prednisone, as this may prevent contralateral eye involvement and stabilize or occasionally partially reverse established visual loss. For patients without cranial ischemic features and without visual risk, 40 mg per day may be appropriate. Patients who present with isolated large-vessel GCA are typically managed with 40–60 mg per day depending on clinical severity.
Low-dose aspirin (75 to 100 mg per day) is recommended as adjunctive therapy based on two observational studies demonstrating a reduction in cranial ischemic events (visual loss, stroke) in GCA patients taking antiplatelet therapy at the time of diagnosis. The mechanism likely involves inhibition of platelet aggregation at sites of vascular inflammation and luminal narrowing. While not the subject of a prospective randomized trial specifically in GCA, the benefit-to-risk ratio in this elderly population is generally favorable.
Corticosteroid tapering must proceed slowly over 12 to 24 months, guided by clinical symptoms and inflammatory markers (ESR, CRP). Relapse during or after taper is common, occurring in approximately 40 to 50% of patients within the first year. Relapse typically manifests as return of headache, jaw claudication, scalp tenderness, or rising inflammatory markers — not always with visual symptoms. Reinstatement or dose escalation of steroids is required for relapse. The cumulative steroid burden in GCA is substantial and causes significant morbidity in this elderly population: diabetes mellitus, osteoporotic fractures, hypertension, cataracts, infections, and avascular necrosis.
Tocilizumab (TCZ), a humanized monoclonal antibody against the interleukin-6 receptor, was approved by the FDA in 2017 specifically for GCA — the first biologic agent approved for this indication — based on the landmark GiACTA trial (Stone et al., NEJM 2017; PMID 28745999). In this phase 3 randomized controlled trial, subcutaneous tocilizumab 162 mg weekly combined with a 26-week prednisone taper achieved sustained remission at 52 weeks in 56% of patients, compared with 14% in the placebo plus standard prednisone taper group, with a significantly lower cumulative corticosteroid dose. Tocilizumab is now considered standard of care for patients who require steroid-sparing therapy, including those with relapsing or refractory disease, those with high-risk steroid toxicity profiles, and many physicians now use it as initial combination therapy. Supportive measures include proton pump inhibitor therapy for gastrointestinal protection, calcium and vitamin D supplementation, bisphosphonate therapy to prevent corticosteroid-induced osteoporosis, and glucose monitoring for steroid-induced hyperglycemia.
GCA–PMR Overlap
Polymyalgia rheumatica and giant cell arteritis are closely related conditions that share pathophysiologic mechanisms and frequently co-occur. Forty to fifty percent of patients with GCA have concurrent PMR — characterized by bilateral aching and pronounced morning stiffness of the shoulder girdles, neck, and hip girdles — either at the time of GCA diagnosis or as a preceding or subsequent manifestation. Conversely, clinically apparent GCA develops in approximately 15 to 20% of patients diagnosed with PMR, and subclinical large-vessel vasculitis detectable on FDG-PET/CT imaging may be present in a higher proportion of "pure PMR" patients without overtly clinical GCA features.
The clinical management implication of this overlap is critical: any patient with a diagnosis of PMR who subsequently develops new headache, jaw claudication, scalp tenderness, visual disturbance, or prominent constitutional symptoms must be urgently evaluated for GCA and started on GCA-dose corticosteroids (prednisone 40 to 60 mg/day, not the lower PMR dose of 15 to 20 mg) while awaiting temporal artery biopsy or imaging. Treating a patient with underlying GCA at only the PMR dose of corticosteroids is inadequate to prevent ischemic complications, including irreversible vision loss. This distinction between the treatment doses for PMR alone versus GCA is one of the most practically important points in rheumatologic emergency management.
Prognosis and Monitoring
Visual loss due to anterior ischemic optic neuropathy is the most devastating acute complication of GCA and is almost always permanent once established. With prompt high-dose corticosteroid treatment initiated before optic nerve infarction occurs, the risk of visual loss is reduced to below 5%; in untreated or delayed-treatment patients, visual loss occurs in 15 to 20%. Contralateral eye involvement — sometimes occurring within days of the first eye — is a particular concern and underscores the urgency of IV methylprednisolone in patients with any established visual loss. Aortic aneurysm, primarily thoracic, represents a significant late complication: patients with GCA have a 2- to 3-fold increased lifetime risk of aortic aneurysm compared with the general population, with most cases presenting 5 to 10 years after the initial GCA diagnosis. Current guidelines recommend surveillance imaging (CT angiography or MRA) of the thoracic aorta at approximately 5 years after diagnosis to detect aortic dilation before rupture or dissection.
The disease typically follows a chronic relapsing-remitting course over 2 to 5 years, after which many patients are able to taper and discontinue corticosteroids entirely. Relapse risk is highest during the first 2 years of taper, particularly when the prednisone dose falls below 10 to 15 mg per day. Inflammatory markers (ESR and CRP) should be monitored regularly during the taper — rising markers in an asymptomatic patient warrant caution and close follow-up, though asymptomatic marker elevation alone does not always mandate dose escalation in a clinically stable patient. Tocilizumab masks the elevation of CRP (a direct downstream effect of IL-6 blockade) and renders CRP unreliable as a disease activity marker in patients on this biologic; clinical symptoms become the primary guide to relapse detection in tocilizumab-treated patients. Long-term steroid-related morbidity — diabetes, osteoporosis, infections, cataracts, and cardiovascular disease — often exceeds the direct morbidity of the vasculitis itself in elderly GCA patients, making steroid-sparing strategies a high priority from the outset of treatment.
References
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371(1):50–57. PMID 24988557
- Stone JH, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377(4):317–328. PMID 28745999
- Dejaco C, et al. 2015 Recommendations for the management of polymyalgia rheumatica. Ann Rheum Dis. 2015;74(10):1799–1807. PMID 26359488
- Hellmich B, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2020;79(1):19–30. PMID 31270110
- Dasgupta B, et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology (Oxford). 2010;49(8):1594–1597. PMID 20371504
- Salvarani C, et al. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med. 2002;347(4):261–271. PMID 12140303
- Muratore F, et al. Large-vessel giant cell arteritis: a cohort study. Rheumatology (Oxford). 2015;54(3):463–470. — Search PubMed
- Hayreh SS, et al. Giant cell arteritis: validity and reliability of various diagnostic criteria. Am J Ophthalmol. 1997;123(3):285–296. — Search PubMed
- Gonzalez-Gay MA, et al. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore). 2005;84(5):269–276. — Search PubMed
- Proven A, et al. Glucocorticoid therapy in giant-cell arteritis: duration and adverse outcomes. Arthritis Rheum. 2003;49(5):703–708. — Search PubMed
- Cid MC, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. 1998;41(1):26–32. — Search PubMed
- Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the management of giant cell arteritis and Takayasu arteritis. Arthritis Care Res (Hoboken). 2021;73(8):1071–1087. — Search PubMed
Connections
- Rheumatology
- Polymyalgia Rheumatica
- Vasculitis
- Polyarteritis Nodosa
- Behçet's Disease
- EGPA
- Neurology
- Lab Tests