Pulmonary Hypertension: History and Discovery
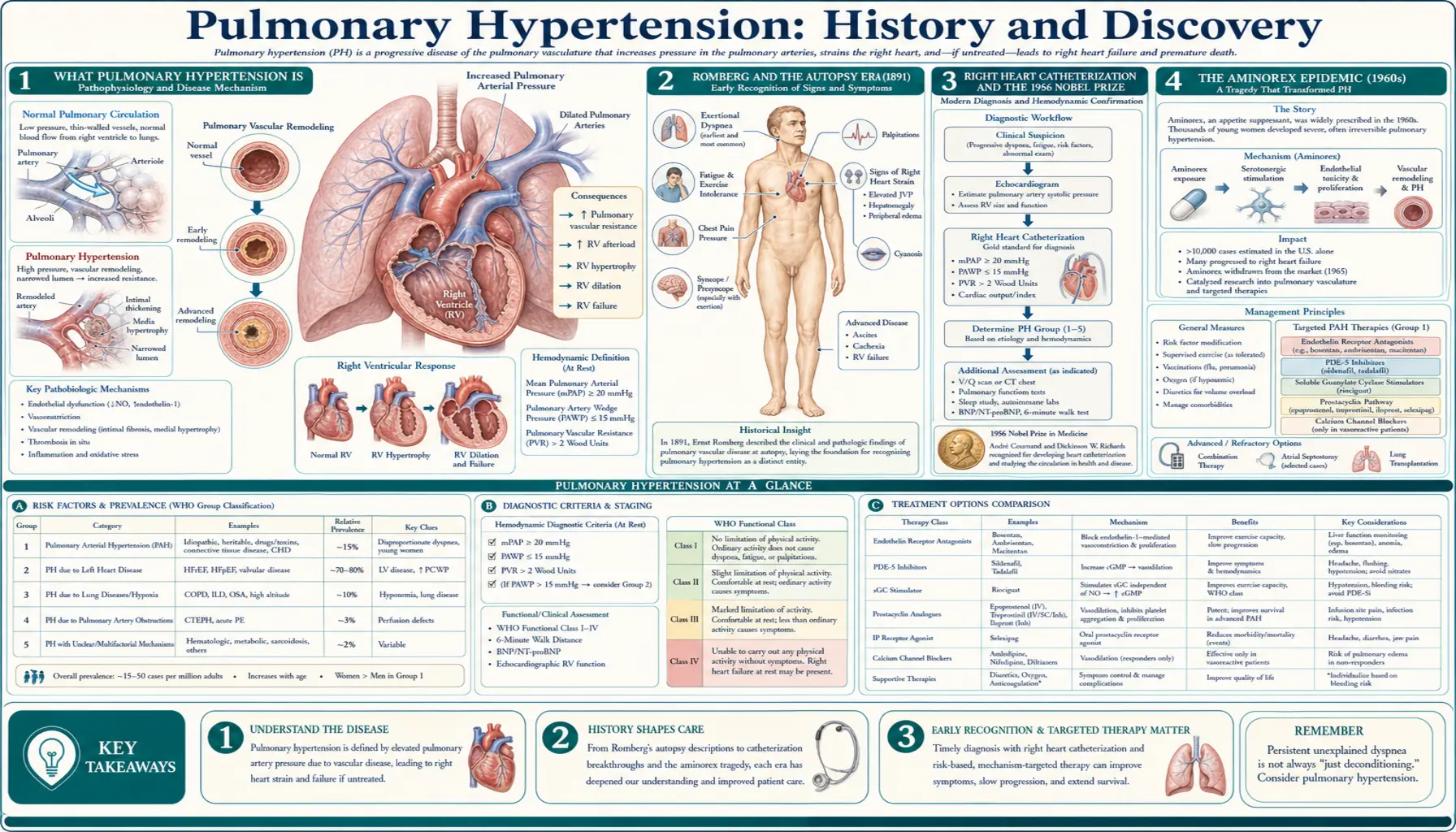
Pulmonary hypertension (PH) is high blood pressure in the arteries that carry blood from the right side of the heart through the lungs. Because those vessels are normally a low-pressure system, even a modest rise in pressure forces the right ventricle to work far harder than it was built to, and over time the strain can lead to right heart failure. For most of medical history the disease was invisible: it could only be recognized at autopsy, where pathologists found thickened, scarred pulmonary arteries with no obvious cause. The German physician Ernst von Romberg gave one of the earliest such descriptions in 1891, calling it "pulmonary vascular sclerosis." Real progress had to wait until doctors could measure pressure inside the living heart — the achievement of right heart catheterization, pioneered by Werner Forssmann (who threaded a catheter into his own heart in 1929) and turned into a clinical tool by André Cournand and Dickinson Richards, work that earned the three men the 1956 Nobel Prize. Two drug-induced "epidemics" — the aminorex appetite-suppressant outbreak in 1960s Europe and the 1990s "fen-phen" diet-drug disaster — proved that medicines could cause PH and galvanized research. From an NIH registry, a modern WHO classification, and a generation of targeted drugs, a once-untreatable disease became, for the first time, something medicine could fight.
Table of Contents
- What Pulmonary Hypertension Is
- Romberg and the Autopsy Era (1891)
- Right Heart Catheterization and the 1956 Nobel Prize
- The Aminorex Epidemic (1960s)
- The NIH Registry and "Primary" Pulmonary Hypertension
- The Fen-Phen Disaster (1990s)
- Classification and the Genetic Breakthrough
- The Era of Targeted Therapy
- Legacy and the Modern Picture
- Research Papers and References
- Connections
- Featured Videos
What Pulmonary Hypertension Is
To follow the history, it helps to picture the plumbing. Blood returning from the body enters the right side of the heart, which pumps it through the pulmonary arteries into the lungs to pick up oxygen. This pulmonary circuit is normally a gentle, low-pressure system — the right ventricle is a thin-walled chamber that never had to push very hard. In pulmonary hypertension, the small arteries of the lungs become narrowed, stiff, or scarred, so the right ventricle must generate much higher pressure to move blood through them. Like any muscle forced to strain, it first thickens and then, eventually, begins to fail.
That single mechanical fact explains why the disease was so hard to discover and so easy to miss. Its symptoms — breathlessness on exertion, fatigue, fainting, swelling of the legs — are shared by dozens of common conditions and point nowhere in particular. There is no rash, no fever, no telltale sign on a routine examination. For the better part of a century the only certain evidence was found after death, in the thickened pulmonary arteries and the enlarged, overworked right ventricle that pathologists saw on the autopsy table. Pulmonary hypertension was, in effect, less a diagnosis than a description of post-mortem findings — and the whole arc of its history is the story of how medicine learned, step by step, first to measure it, then to classify its many causes, and finally to treat it.
Romberg and the Autopsy Era (1891)
The recorded history of pulmonary hypertension usually begins with the German physician Ernst von Romberg, who in 1891 published an account of a patient whose lungs, examined after death, showed strikingly thickened and damaged pulmonary arteries. Romberg described the abnormality as pulmonary vascular sclerosis — a hardening of the lung's blood vessels — and his report is widely cited as the first clear description of what we now call pulmonary hypertension. He had identified the disease's anatomical signature without any means of measuring the pressure that produced it.
That limitation defined the entire early era. With no instrument able to record pressure inside the chambers of the heart or the pulmonary arteries of a living person, physicians could only infer that the pressure must have been high from the remodeling they found at autopsy. Over the following decades a handful of investigators added further pathological descriptions, but the disease remained, in practice, a diagnosis made only after a patient had died. Romberg's contribution was therefore less a single discovery than the opening of a long question: here was a deadly narrowing of the lung's arteries, plainly visible to the pathologist, that medicine could neither detect in time nor explain. Answering it would require a way to reach inside the living heart.
Right Heart Catheterization and the 1956 Nobel Prize
The tool that finally made pulmonary hypertension a living diagnosis was right heart catheterization — threading a thin tube through a vein, into the right side of the heart, and onward into the pulmonary artery to measure pressure directly. Its origin is one of the most famous acts of self-experimentation in medicine. In 1929, a young German surgical trainee named Werner Forssmann, working in Eberswalde near Berlin and convinced a catheter could safely be passed into the human heart, inserted a urinary catheter into a vein in his own arm, advanced it into his right atrium, and then walked to the radiology department to document its position by X-ray. His superiors regarded the feat as reckless; criticized and discouraged, Forssmann eventually left cardiology for a career in urology.
His idea was not lost. At Bellevue Hospital and Columbia University in New York, the physiologists André Frederic Cournand and Dickinson W. Richards recognized that cardiac catheterization could become a precise instrument for studying the heart and lungs. Through the late 1930s and into the 1940s they refined the technique into a reliable clinical and research procedure, using it to measure pressures and blood flow in the human heart in ways never before possible. For the first time, the pressure in the pulmonary arteries of a living patient could be read off directly — transforming pulmonary hypertension from an autopsy finding into a measurable, diagnosable condition.
In 1956 the achievement was recognized at the highest level: the Nobel Prize in Physiology or Medicine was awarded jointly to Cournand, Forssmann, and Richards "for their discoveries concerning heart catheterization and pathological changes in the circulatory system." The award is notable for honoring both the audacious originator of the idea and the two investigators who made it medically useful. Right heart catheterization remains, to this day, the definitive test that confirms pulmonary hypertension and measures its severity.
The Aminorex Epidemic (1960s)
The first great clue that pulmonary hypertension could be caused — not merely found — came from an unexpected and tragic source: a diet pill. Aminorex (aminorex fumarate), an amphetamine-like appetite suppressant, was introduced in parts of central Europe in the mid-1960s; it was registered as an anorexigen around 1965 and sold over the counter in Switzerland, Austria, and West Germany. Within a few years, physicians in those three countries noticed a sharp and frightening rise in cases of severe pulmonary hypertension in otherwise healthy people — an outbreak so far beyond the normal background rate that it amounted to an epidemic.
Careful epidemiological detective work tied the surge to aminorex: the number of cases rose and fell with the drug's sales, the affected patients were overwhelmingly people who had taken it, and the geography of the outbreak matched the countries where it was marketed. After the drug was withdrawn around 1968, the wave of new cases subsided. Only a small fraction of those who took aminorex — on the order of one or two percent — developed the disease, which is why the connection took time to recognize, but for those affected the consequences were frequently fatal in an era with no effective treatment.
The aminorex episode permanently changed how physicians thought about the disease. It established, for the first time, that an ingested drug could trigger pulmonary hypertension, and it pointed suspicion toward the appetite-suppressant class in particular — a lesson that would prove bitterly prophetic three decades later.
The NIH Registry and "Primary" Pulmonary Hypertension
For decades, pulmonary hypertension with no identifiable cause was labeled primary pulmonary hypertension (PPH) — "primary" meaning it arose on its own, as opposed to "secondary" PH caused by another known disease such as heart or lung disorders. Because PPH was rare and uniformly studied at scattered centers, no one had a clear, systematic picture of who got it, how it behaved, or how long patients survived. In the United States, the National Institutes of Health set out to fix that.
The NIH Registry on Primary Pulmonary Hypertension began enrolling patients in 1981, gathering data from dozens of medical centers on patients diagnosed by uniform criteria. The registry's findings, reported in the late 1980s, painted the first reliable portrait of the disease: it struck adults at an average age in the mid-thirties, affected women considerably more often than men, and most commonly announced itself with breathlessness, fatigue, and fainting. Crucially, the registry also documented how grim the outlook then was — in the era before targeted therapy, median survival after diagnosis was measured in only a few years — and produced an equation, based on pressures measured at catheterization, to estimate a given patient's prognosis.
The NIH Registry mattered for two reasons. It gave clinicians and researchers a shared, evidence-based baseline against which every future treatment could be measured, and its sobering survival data made unmistakably clear how badly new therapies were needed. When effective drugs finally arrived in the 1990s, it was against this benchmark that their life-extending benefit was proven.
The Fen-Phen Disaster (1990s)
The lesson of aminorex was relearned, on a far larger scale, in the United States in the 1990s. A wildly popular weight-loss combination known as "fen-phen" — the appetite suppressant fenfluramine (and its close relative dexfenfluramine) paired with phentermine — was prescribed to millions of people. Building directly on the aminorex experience, an international case-control investigation, the International Primary Pulmonary Hypertension Study (IPPHS), reported in the New England Journal of Medicine in 1996 that use of these appetite-suppressant drugs sharply increased the risk of primary pulmonary hypertension, with the risk climbing the longer the drugs were taken.
Then, in 1997, the danger widened. Physicians at the Mayo Clinic described an unusual heart-valve disease in women who had taken fen-phen, and similar reports quickly accumulated. Faced with mounting evidence that fenfluramine and dexfenfluramine were linked both to pulmonary hypertension and to valve damage, the U.S. Food and Drug Administration moved against them, and the drugs were withdrawn from the market in September 1997. The episode triggered one of the largest pharmaceutical liability sagas in American history, ultimately involving tens of thousands of claimants.
For the field of pulmonary hypertension, the fen-phen disaster was a grim confirmation and a powerful spur. It proved beyond doubt that drugs could cause the disease, drew enormous public and scientific attention to a once-obscure condition, and accelerated the search for both its underlying biology and its treatment. The appetite-suppressant epidemics — aminorex and fen-phen alike — turned out to be tragic natural experiments that taught medicine a great deal about how the pulmonary arteries are injured.
Classification and the Genetic Breakthrough
As understanding grew, so did the need to organize a disease with many different causes into a coherent scheme. The first attempt came at a World Health Organization symposium in Geneva in 1973, which drew the basic line between "primary" pulmonary hypertension (no identifiable cause) and "secondary" PH (caused by another condition). That simple division served for a generation but could not capture the disease's true diversity. At a second WHO world symposium held in Evian, France, in 1998, experts replaced it with a clinical classification that grouped patients by shared mechanisms, clinical features, and treatment responses — the foundation of the modern five-group WHO classification used and refined today (Group 1 pulmonary arterial hypertension; Group 2 due to left heart disease; Group 3 due to lung disease and low oxygen; Group 4 from chronic blood clots; and Group 5, miscellaneous). In step with this framework, the old term "primary pulmonary hypertension" was retired in favor of idiopathic pulmonary arterial hypertension (IPAH).
The second great advance of this period was genetic. Clinicians had long known that PPH sometimes ran in families, behaving as an inherited trait. In 2000, researchers tracked that inheritance to its source, reporting that mutations in a gene called BMPR2 — which encodes a receptor in the transforming growth factor-beta (TGF-β) signaling family — cause heritable pulmonary arterial hypertension. The familial form had been mapped to a region of chromosome 2 (the locus designated PPH1), and BMPR2 was identified as the responsible gene.
The BMPR2 discovery reframed pulmonary arterial hypertension as, in part, a disease of disordered cell signaling and growth in the walls of the small lung arteries — explaining why those vessels thicken and remodel — and gave scientists a concrete molecular pathway to study. The immediate revolution in patient care, however, came from a different direction entirely: targeted drugs.
The Era of Targeted Therapy
Until the mid-1990s, there were no drugs designed specifically for pulmonary hypertension. Doctors borrowed general blood-pressure medicines and vasodilators that acted on arteries throughout the whole body, with limited benefit and considerable side effects. The turning point came in December 1995, when the FDA approved continuous intravenous epoprostenol (a synthetic form of prostacyclin, marketed as Flolan) for primary pulmonary hypertension. Prostacyclin is a natural substance that relaxes and widens blood vessels and discourages clotting; in a clinical trial, continuous infusion of epoprostenol improved symptoms, hemodynamics, and — remarkably for this disease — survival. It was the first targeted therapy for the condition, albeit a demanding one, requiring a permanent indwelling catheter and a continuous pump.
Two further drug classes followed, each aimed at a specific molecular pathway in the diseased pulmonary arteries. In November 2001, bosentan (Tracleer) became the first endothelin receptor antagonist approved for pulmonary arterial hypertension — an oral drug that blocks endothelin, a powerful natural vessel-constrictor that is overactive in the disease. Then, in June 2005, sildenafil (marketed for this use as Revatio) was approved as a phosphodiesterase-5 (PDE5) inhibitor; the same molecule famous under another brand name relaxes the pulmonary arteries by preserving a signaling molecule (cyclic GMP) that keeps the vessels open. Together, prostacyclins, endothelin receptor antagonists, and PDE5 inhibitors gave clinicians, for the first time, oral and infused options that could be combined.
The arrival of these three pathways changed the disease's whole trajectory. A condition that the NIH Registry had shown to be fatal within a few years for many patients became one that could often be managed for far longer, with meaningfully better symptoms, exercise capacity, and survival. None of these drugs is a cure, and pulmonary arterial hypertension remains serious and demanding to treat, but the contrast with the untreatable disease of the 1980s is profound.
Legacy and the Modern Picture
The history of pulmonary hypertension is, in a sense, a history of seeing. For most of the era after Romberg's 1891 description, the disease was visible only to the pathologist; the catheter let physicians see it in the living; the registries let them see it as a population; genetics let them see it at the level of a single gene; and targeted drugs, at last, let them change what they saw. Each of those steps converted a hidden, hopeless condition into one that is detected, classified, and treated.
Two themes deserve emphasis. The first is the central, recurring role of drug-induced epidemics: aminorex in 1960s Europe and fen-phen in 1990s America were human tragedies, but they also produced some of the clearest evidence that the pulmonary arteries can be specifically injured, and they accelerated the science that followed. The second is the speed of the modern transformation — the leap from "no specific treatment exists" in 1994 to three distinct, mechanism-based drug classes within little more than a decade is one of the more dramatic therapeutic turnarounds in pulmonary medicine.
This page records the historical arc of discovery; it is not medical advice. Pulmonary hypertension remains a serious diagnosis that requires specialist evaluation — today centered on echocardiography for screening and right heart catheterization for confirmation. Anyone with unexplained breathlessness, fatigue, fainting, or leg swelling should be assessed by a clinician, because the difference early diagnosis and modern therapy now make is exactly the difference this history describes.
Research Papers and References
The references below combine landmark peer-reviewed papers in the history of pulmonary hypertension — with real DOIs or PubMed identifiers where they are well established — with curated PubMed topic-search links into the broader historical and clinical literature. Romberg's 1891 report and the 1956 Nobel award are described in the article as historical events and are documented in the review articles cited here. Each link opens in a new tab at its source.
- Forssmann W. Die Sondierung des rechten Herzens (Catheterization of the right heart). Klinische Wochenschrift. 1929;8(45):2085-2087. — doi:10.1007/BF01875120
- The Nobel Prize in Physiology or Medicine 1956 — Cournand, Forssmann, and Richards, "for their discoveries concerning heart catheterization and pathological changes in the circulatory system." — NobelPrize.org — 1956 Physiology or Medicine
- Gurtner HP. Aminorex and pulmonary hypertension. A review. Cor et Vasa. 1985;27(2-3):160-171. — Search PubMed
- Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension: a national prospective study. Annals of Internal Medicine. 1987;107(2):216-223. — doi:10.7326/0003-4819-107-2-216
- D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Annals of Internal Medicine. 1991;115(5):343-349. — doi:10.7326/0003-4819-115-5-343
- Abenhaim L, Moride Y, Brenot F, et al; International Primary Pulmonary Hypertension Study Group. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. New England Journal of Medicine. 1996;335(9):609-616. — doi:10.1056/NEJM199608293350901
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. New England Journal of Medicine. 1997;337(9):581-588. — doi:10.1056/NEJM199708283370901
- Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. New England Journal of Medicine. 1996;334(5):296-301. — doi:10.1056/NEJM199602013340504
- Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. New England Journal of Medicine. 2002;346(12):896-903. — doi:10.1056/NEJMoa012212
- Galiè N, Ghofrani HA, Torbicki A, et al; Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. New England Journal of Medicine. 2005;353(20):2148-2157. — doi:10.1056/NEJMoa050010
- The International PPH Consortium; Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nature Genetics. 2000;26(1):81-84. — doi:10.1038/79226
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. European Respiratory Journal. 2019;53(1):1801913. — doi:10.1183/13993003.01913-2018
- History and evolution of pulmonary hypertension classification and nomenclature (WHO Geneva 1973; Evian 1998) PubMed: pulmonary hypertension classification history
- Werner Forssmann and the history of cardiac catheterization PubMed: Forssmann history of cardiac catheterization
External Authoritative Resources
- NHLBI (NIH) — Pulmonary Hypertension
- NobelPrize.org — Nobel Prize in Physiology or Medicine 1956
- PubMed — History of pulmonary hypertension
Connections
- Pulmonology
- Pulmonary Hypertension (main article)
- Heart Failure
- Pulmonary Embolism
- Interstitial Lung Disease
- Hypertension
- All Conditions