Hirschsprung's Disease (Congenital Aganglionic Megacolon)
- Overview and Epidemiology
- Genetics and Molecular Pathogenesis
- Anatomy of Aganglionosis
- Pathophysiology and Clinical Presentation
- Diagnosis
- Surgical Treatment
- Postoperative Outcomes and Complications
- HAEC Recognition and Management
- Key Research Papers
- Connections
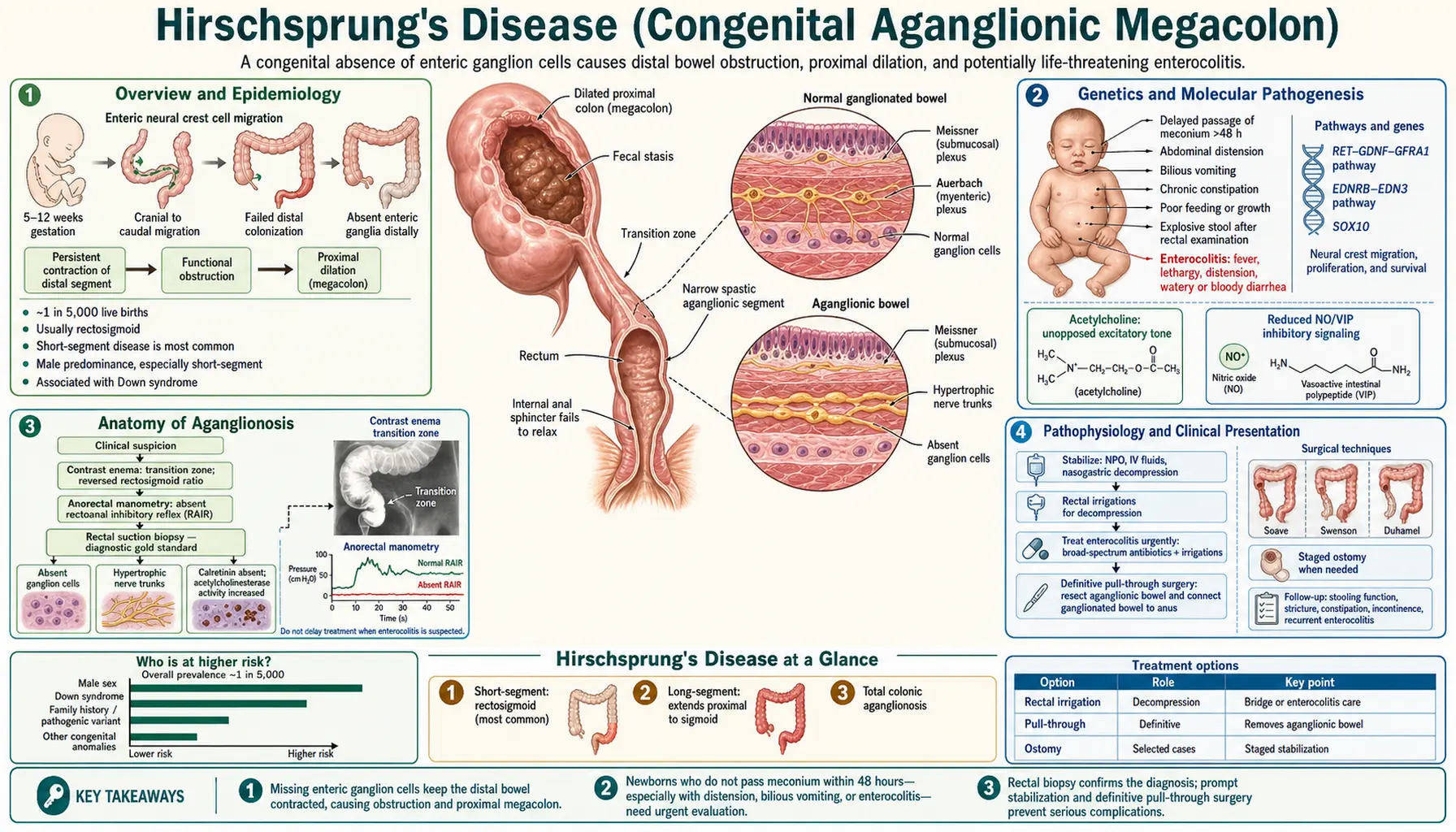
Overview and Epidemiology
Hirschsprung's disease (HD), also called congenital aganglionic megacolon, is a congenital defect characterized by the complete absence of ganglion cells (aganglionosis) in the distal bowel. It results from failure of neural crest cells to migrate fully into the hindgut during embryonic development — a process that normally completes between weeks 5 and 12 of gestation. Without ganglion cells, the affected segment of bowel cannot relax or coordinate peristalsis, creating a functional obstruction that causes the normally-innervated proximal bowel to balloon outward: the megacolon.
HD occurs in approximately 1 in 5,000 live births worldwide and is one of the most common causes of neonatal intestinal obstruction. The condition affects males far more often than females in its most common (short-segment) form, with a male-to-female ratio of approximately 4:1. This ratio narrows toward 1:1 in long-segment disease, which is more uniformly distributed across sexes and carries a higher genetic burden.
Most cases are sporadic, but familial clustering is well recognized. Important disease associations include:
- Down syndrome (trisomy 21): Approximately 2–10% of HD patients have Down syndrome; conversely, roughly 4% of children with Down syndrome have HD. Any newborn with Down syndrome who fails to pass meconium within 48 hours requires urgent evaluation for HD.
- Multiple endocrine neoplasia type 2 (MEN2): Caused by RET proto-oncogene mutations — the same gene most commonly mutated in familial HD. Children with HD should be screened for RET mutations; a pathogenic RET variant in a HD patient warrants genetic counseling for the family regarding medullary thyroid carcinoma risk.
- Waardenburg syndrome type 4 (Shah-Waardenburg): Combines features of Waardenburg syndrome (hearing loss, pigmentation abnormalities) with HD, due to mutations in EDNRB, EDN3, or SOX10.
Genetics and Molecular Pathogenesis
HD is a complex genetic disorder in which multiple genes, each with incomplete penetrance, contribute to disease risk. Understanding the genetics is increasingly important as genetic counseling, cancer surveillance, and family planning decisions depend on identifying the causative variant.
RET proto-oncogene (chromosome 10q11.21) is the major susceptibility gene. RET encodes a receptor tyrosine kinase expressed on enteric neural crest cells (NCC). Its signaling is driven by the GDNF (glial cell line-derived neurotrophic factor) / GFRα1 ligand complex. RET activation promotes NCC survival, proliferation, and caudal migration through the developing gut. Loss-of-function mutations in RET — numbering in the hundreds — impair this process and are found in:
- ~50% of familial HD cases
- ~15–20% of sporadic short-segment HD
- ~70–80% of long-segment and total colonic HD
The penetrance of RET mutations is incomplete and sex-influenced — males with a pathogenic RET variant have a much higher lifetime risk of HD than females, explaining the strong male predominance in short-segment disease.
Other HD-associated genes and their pathways:
- EDNRB (endothelin receptor B) and EDN3 (endothelin-3): The endothelin signaling axis regulates NCC differentiation and migration independently of RET. EDNRB mutations are the second most common cause of familial HD and account for the majority of Shah-Waardenburg syndrome cases. Notably, heterozygous EDNRB mutations in isolation have low penetrance, but homozygous or compound heterozygous mutations cause the full Waardenburg-Shah phenotype.
- SOX10: A transcription factor essential for NCC multipotency; mutations cause Shah-Waardenburg syndrome and isolated long-segment HD.
- PHOX2B: Mutations cause congenital central hypoventilation syndrome (Ondine's curse), which can be associated with HD — a combination called Haddad syndrome.
- ZFHX1B (SIP1/ZEB2): Mutations cause Mowat-Wilson syndrome (HD + intellectual disability + Hirschsprung).
Inheritance patterns vary by segment length and gene involved. Short-segment HD behaves as a low-penetrance dominant trait with strong sex bias; long-segment HD often has higher heritability and may follow more Mendelian patterns when a single high-penetrance gene is involved. Genetic testing panels for HD should include at minimum RET, EDNRB, EDN3, and SOX10 in familial cases or cases with associated anomalies.
Anatomy of Aganglionosis
Understanding where the aganglionic segment begins and how far it extends is fundamental to both diagnosis and surgical planning. There is one anatomical invariant: the aganglionic segment always begins at the internal anal sphincter (IAS) and extends proximally to a variable length. There is no form of HD in which the distal rectum is spared. This is why a normal rectal biopsy at the appropriate level essentially excludes HD.
The normally-innervated bowel contains two ganglionated plexuses:
- Myenteric (Auerbach's) plexus: Located between the circular and longitudinal muscle layers; controls coordinated peristaltic contractions.
- Submucosal (Meissner's) plexus: Located in the submucosa; controls secretion, absorption, and local reflex arcs including the rectoanal inhibitory reflex (RAIR).
In HD, both plexuses are absent from the aganglionic segment. This total absence of inhibitory neuronal input means the IAS and the aganglionic bowel remain in a state of tonic contraction, incapable of the coordinated relaxation that normally allows stool to pass.
The transition zone (TZ) — the region between the aganglionic distal bowel and the normally-ganglionated proximal bowel — shows irregular and hypoganglionic (sparse, immature) ganglion cells. Pathologically, the TZ is characterized by hypertrophied, acetylcholinesterase-positive nerve trunks in the submucosa. The TZ is diagnostically important and surgically critical: pull-through procedures must place the anastomosis well above the TZ in fully-ganglionated bowel.
Classification by segment length:
- Short-segment HD (80% of cases): Aganglionosis confined to the rectosigmoid region, distal to the splenic flexure. This is the classic form.
- Long-segment HD (approximately 15%): Aganglionosis extends proximal to the sigmoid colon into the descending colon or further. Genetic burden is higher; more likely to have an identifiable RET mutation.
- Total colonic aganglionosis (TCA, approximately 5%): The entire colon is aganglionic with variable involvement of the distal small bowel. Diagnosis is often delayed because the typical contrast enema appearance is different — the colon may appear small-caliber throughout without a clear transition zone. Requires specialized surgical approaches (ileoanal or extended pull-through).
- Total intestinal aganglionosis (TIA, <1%): Extremely rare; involves most of the small intestine. Often incompatible with long-term survival without intestinal transplantation.
- Ultrashort-segment HD: A controversial entity in which aganglionosis is limited to the most distal internal anal sphincter region. Diagnosis and management are debated; some experts classify it separately from classic HD.
Pathophysiology and Clinical Presentation
The core pathophysiological defect is straightforward: without ganglion cells, the distal bowel cannot relax on command. Stool that reaches the aganglionic segment encounters a tonically contracted segment that acts as a mechanical valve. Proximal bowel distends as stool accumulates above the obstruction — hence megacolon. The rectoanal inhibitory reflex (RAIR) — the normal reflex relaxation of the IAS when the rectum is distended — is absent in HD. This is the physiological basis for anorectal manometry as a diagnostic test.
Neonatal presentation (the majority of HD is diagnosed in the newborn period):
- Failure to pass meconium within the first 48 hours of life is present in approximately 99% of HD patients and is the most sensitive clinical sign. In a healthy term neonate, 94% pass meconium within the first 24 hours; virtually all do so by 48 hours. Any term newborn who has not passed meconium by 48 hours requires immediate evaluation for HD and other causes of intestinal obstruction.
- Abdominal distension: Progressive, often tense, with a tympanitic abdomen.
- Bilious vomiting: Indicates obstruction at or distal to the duodenal ampulla of Vater.
- Explosive decompression with rectal stimulation: One of the most clinically useful findings — when a digital rectal exam or rectal thermometer is inserted, there is often an explosive release of gas and meconium as the functional obstruction is temporarily bypassed. This "squirt sign" provides temporary relief and is both diagnostic and therapeutic in the initial evaluation.
Presentation in older children (milder cases missed in the neonatal period, or ultrashort-segment HD):
- Constipation since birth: This is the key distinguishing feature from functional constipation. A child with HD has never had normal stooling — the difficulty began from day one. Parents of children with functional constipation can usually identify a time when their child stooled normally before the problem started (toilet training, dietary change, painful anal fissure). HD patients have no such history.
- Abdominal distension and failure to thrive.
- Ribbon-like or small-caliber stools: The aganglionic segment acts as a narrow, non-dilating stricture, so any stool that does pass is forced through a narrow channel.
- Paradoxical diarrhea: Overflow incontinence around an impacted stool bolus can mimic diarrhea and lead to misdiagnosis as infectious gastroenteritis.
Hirschsprung-Associated Enterocolitis (HAEC) is the most feared acute complication of HD, responsible for the majority of HD-related deaths. HAEC occurs in 15–50% of patients with HD, most commonly in the first months of life before diagnosis (pre-operative HAEC) but also after a technically successful pull-through procedure (post-operative HAEC). Pathogenesis involves bacterial overgrowth in the massively distended proximal bowel, disruption of the mucus barrier, bacterial translocation, and an overwhelming inflammatory cascade. Clinical presentation: fever + explosive foul-smelling diarrhea + severe abdominal distension. The "squirt sign" on rectal exam — explosive discharge when a rectal tube is inserted — is pathognomonic. Without prompt treatment, HAEC progresses to septic shock and colonic perforation. HAEC is a surgical emergency requiring immediate hospitalization, IV antibiotics, and rectal irrigations.
Diagnosis
The diagnosis of HD rests on three complementary modalities. No single test is 100% sensitive and specific; the clinical picture must integrate all available data. In a newborn with failure to pass meconium and abdominal distension, diagnostic workup should proceed urgently.
Rectal suction biopsy — the gold standard: A suction biopsy device is used to take 3 small mucosal and submucosal specimens at 2, 3, and 4 cm above the dentate line (sampling below 2 cm is unreliable because there is a normal physiologic hypoganglionic zone immediately above the dentate line in all infants). The pathologist evaluates:
- Absence of ganglion cells in both the submucosal and myenteric plexuses — required criterion for HD diagnosis.
- Hypertrophied nerve trunks on acetylcholinesterase (AChE) histochemistry — in the aganglionic segment, AChE-positive parasympathetic nerve fibers are dense and hypertrophic (paradoxically, these nerve fibers INCREASE in the absence of ganglion cells, as if attempting to compensate). This is the key positive finding that distinguishes HD from other causes of neonatal obstruction.
Rectal suction biopsy can be performed at the bedside without general anesthesia in neonates and young infants. A full-thickness biopsy under general anesthesia is reserved for cases where suction biopsy is technically inadequate or histologically inconclusive. Calretinin immunostaining is increasingly used as an adjunct — calretinin-positive ganglion cells and nerve fibers are present in normal bowel but absent in aganglionic bowel.
Contrast enema: A water-soluble contrast enema (barium avoided in acutely ill neonates due to perforation risk) is performed without prior rectal preparation (which would wash out the transition zone). The lateral view is the most informative. Classic findings:
- Transition zone: The aganglionic segment appears relatively narrow; the normally-innervated proximal bowel is dilated. This creates a "funnel" or transition zone on lateral view, visible in approximately 70% of cases. Counterintuitively, the narrow segment is the diseased one — it is narrowed because it cannot relax, while the proximal normal bowel has dilated in response to the obstruction.
- 24-hour delayed film: Retained contrast after 24 hours indicates poor evacuation, supporting the diagnosis.
- Total colonic HD caveat: When the entire colon is aganglionic, there may be no obvious transition zone; instead the entire colon appears small-caliber (microcolon). This presentation can be confused with meconium ileus or other neonatal obstructions.
Anorectal manometry: Measures the RAIR by inflating a small rectal balloon and recording internal anal sphincter pressure. In normal individuals (even preterm infants beyond ~26 weeks), rectal distension causes reflex relaxation of the IAS. In HD, the RAIR is absent — the IAS fails to relax, or may paradoxically contract. Anorectal manometry has sensitivity and specificity exceeding 90% for HD in experienced hands and is particularly useful for evaluating older children with constipation and for post-operative surveillance. It requires technical expertise and is less reliable in very young neonates.
Key clinical rule: Any child with constipation that began at or near birth — without an identifiable trigger and without a period of normal stooling — should be evaluated for HD until proven otherwise. Functional constipation in a true newborn (the first month of life) is a diagnosis of exclusion.
Surgical Treatment
The definitive treatment of HD is surgical removal of the aganglionic segment with a pull-through procedure — mobilizing normally-ganglionated proximal bowel and bringing it down to the anal verge to create a new, functional anastomosis. The goal is to restore propulsive peristalsis to the rectal outlet while preserving the voluntary external anal sphincter mechanism responsible for continence.
Historical procedures — understanding these helps explain why multiple modern approaches exist and what each prioritizes:
- Swenson procedure (1948): The original pull-through, developed by Orvar Swenson. Transabdominal mobilization of the aganglionic rectum down to just above the dentate line, followed by transperineal primary anastomosis. Established the proof of concept but required a two-layer perineal anastomosis close to the sphincters, with risk of nerve damage.
- Duhamel procedure (1956): A side-to-side colorectal anastomosis in which normally-ganglionated bowel is brought behind the aganglionic rectum. The anterior wall of the aganglionic rectum is left in place as a pouch; the posterior wall is anastomosed to the pulled-through colon. Technically simpler; still used for long-segment and total colonic HD.
- Soave endorectal pull-through (1964): The rectal mucosa is stripped from its muscular cuff (an endorectal dissection), and normally-ganglionated bowel is pulled through the preserved aganglionic muscular cuff down to the perineum. Avoids circumferential perineal dissection; risk of retained aganglionic muscle cuff causing obstruction if the cuff is too long.
Modern laparoscopic and transanal approaches:
- Transanal endorectal pull-through (TEPT): The preferred approach for most short-segment HD in experienced pediatric surgical centers. The entire operation is performed through the anus — no abdominal incision required. The rectal mucosa is incised circumferentially just above the dentate line, the endorectal dissection proceeds cephalad within the muscular cuff, and normally-ganglionated bowel is pulled through and anastomosed to the dentate line. Benefits: no abdominal scars, faster recovery, equivalent functional outcomes to open procedures. Requires confirmed intraoperative frozen section histology to ensure the pulled-through bowel contains normal ganglion cells.
- Laparoscopic-assisted pull-through: For long-segment HD where purely transanal mobilization is insufficient — laparoscopy mobilizes the proximal bowel while the transanal pull-through completes the anastomosis.
Timing and staging:
- Single-stage pull-through: Now possible and preferred in most stable neonates and infants; performed within the first weeks to months of life.
- Staged approach (colostomy first): Reserved for acutely ill or septic infants (particularly those presenting with HAEC), extreme prematurity, or complex long-segment disease. A diverting colostomy is placed at a site confirmed to contain ganglion cells (intraoperative frozen section); definitive pull-through is performed at 3–6 months of age when the child is stable.
Pre-operative rectal irrigations: For infants awaiting pull-through, saline irrigations via a rectal catheter are performed twice daily to decompress the obstructed bowel and reduce the risk of HAEC. Parents are taught the technique for home use; this intervention significantly reduces pre-operative morbidity.
Postoperative Outcomes and Complications
The long-term prognosis for HD treated at experienced pediatric surgical centers is generally excellent. The majority of children with short-segment HD achieve socially acceptable fecal continence and lead normal lives. However, a meaningful minority experience ongoing bowel dysfunction that requires continued management, and families should be counseled realistically about the trajectory of improvement — which often extends well into adolescence.
Functional outcomes:
- Approximately 85–90% of children with short-segment HD achieve fecal continence by school age.
- Improvement continues through childhood and adolescence; outcomes at age 10 are meaningfully better than at age 5.
- Total colonic HD has a higher morbidity burden: chronic diarrhea from reduced colonic water absorption, electrolyte losses, and ongoing risk of HAEC are management challenges.
Common postoperative complications:
- Hirschsprung-Associated Enterocolitis (HAEC): The most important post-operative complication; occurs in 15–25% of patients even after technically successful pull-through. The risk persists for years. Post-operative HAEC has similar pathogenesis and management to pre-operative HAEC (IV antibiotics, rectal irrigations, hospitalization). Families of post-operative HD patients must be educated to recognize early HAEC signs and seek emergency care immediately.
- Obstructive symptoms / constipation: Affects 10–30% post-operatively. Causes include: (1) residual aganglionic segment left at the anastomosis (requires redo pull-through); (2) anastomotic stricture (managed with serial dilation); (3) internal anal sphincter achalasia (aganglionic IAS that was not removed — managed with posterior myectomy or intrasphincteric botulinum toxin injection); (4) motility disorder in the pulled-through segment.
- Soiling and fecal incontinence: Affects 25–30% at any given time. Causes include sphincter injury during pull-through, inadequate residual rectal reservoir, or hypersensitive/hyperactive rectum. Tends to improve with age. Bowel management programs (including dietary modification, laxatives, or antegrade irrigation via appendicostomy in severe cases) are effective.
- Anastomotic stricture: Narrowing at the anastomotic site requiring regular dilation; more common with technically challenging pull-throughs or post-operative complications.
Quality of life: Long-term studies consistently show that the majority of HD patients report good quality of life (QoL). QoL scores continue to improve through adolescence. Psychosocial support — addressing body image, school performance, and peer relationships in children with soiling — is an integral part of long-term HD management. Adolescents who had HD as infants benefit from transition programs that prepare them to manage their own bowel health as adults.
HAEC Recognition and Management
Hirschsprung-Associated Enterocolitis (HAEC) is the most dangerous complication of HD and the leading cause of HD-related death. Every parent of a child with HD — diagnosed or suspected — must know how to recognize it and when to seek emergency care. Every clinician caring for a neonate with abdominal distension and fever must consider HAEC before assuming infectious gastroenteritis.
Pathogenesis: The obstructed, distended proximal bowel provides a reservoir for bacterial overgrowth. The mucus barrier — which normally prevents bacterial attachment to the colonic epithelium — is disrupted in HD patients due to altered mucin composition and goblet cell function. Bacterial translocation triggers a cascade of epithelial injury, cytokine release, and systemic inflammation. The organisms most commonly implicated include Clostridium difficile, Cryptosporidium, and aerobic gram-negative bacteria, though HAEC is often polymicrobial. The exact initiating event varies; HAEC can recur even after resolution of the primary obstruction via pull-through.
Clinical recognition — the HAEC triad:
- Fever (often high-grade, >38.5°C / 101.3°F)
- Severe abdominal distension (tense, tympanitic, often with visible peristaltic waves)
- Explosive foul-smelling diarrhea or "squirt sign" — when a rectal tube or gloved finger is inserted, there is an explosive release of liquid stool and gas under pressure. This decompressive squirt is pathognomonic for HAEC in the setting of known or suspected HD.
Additional features: lethargy, poor feeding, vomiting, signs of dehydration, and in severe cases, peritoneal signs suggesting impending perforation. HAEC can progress to septic shock within hours.
Grading: The Pastor HAEC score (based on history, examination, and radiographic findings) stratifies severity and guides urgency of intervention. Radiographs of the abdomen may show dilated loops of bowel, air-fluid levels, pneumatosis intestinalis (gas within the bowel wall — a sign of mucosal ischemia and impending perforation), or free air indicating perforation.
Acute management:
- Immediate hospitalization — HAEC is a medical/surgical emergency; outpatient management is not appropriate for established HAEC.
- IV antibiotics: Coverage must include anaerobes and gram-negative organisms — typically metronidazole plus a broad-spectrum beta-lactam (e.g., piperacillin-tazobactam) or third-generation cephalosporin. Duration depends on clinical response, typically 5–7 days.
- Rectal irrigations — the cornerstone of acute HAEC management: Saline irrigations (10–20 mL/kg of normal saline per irrigation, via a large-bore rectal catheter inserted 5–10 cm above the dentate line) performed every 4–6 hours to mechanically decompress the obstructed bowel and wash out bacterial toxins. Each irrigation should produce a large return of liquid stool and gas; inadequate return is a warning sign of very high obstruction or impending perforation.
- IV fluid resuscitation: Isotonic fluid boluses for hemodynamic instability; ongoing maintenance with electrolyte monitoring (hyponatremia and hypokalemia are common from diarrheal losses).
- Bowel rest and nasogastric decompression: NPO, NG tube on continuous suction for severe cases.
- Surgical escalation: Failure to respond to 24–48 hours of medical management, worsening distension, peritoneal signs, or free air on radiograph mandates emergency surgical consultation for diverting colostomy or, in the most severe cases, emergency pull-through or bowel resection.
Prevention and long-term management of recurrent HAEC: Families of all HD patients — both pre-operative and post-operative — should be taught rectal irrigation technique for home use when HAEC risk is elevated. Post-operative patients at high risk for recurrent HAEC (those with prior episodes, total colonic HD, or Down syndrome) benefit from regular prophylactic irrigations and/or oral probiotics (early data suggest Lactobacillus-based preparations may reduce HAEC frequency). Close surveillance and a low threshold for IV antibiotics at the first sign of recurrence are essential.
Key Research Papers
- Search PubMed — Comprehensive review of the molecular genetics of HD covering RET, EDNRB, EDN3, SOX10, and other loci; discusses penetrance, phenotype-genotype correlations, and genetic counseling implications.
- Search PubMed — Detailed surgical review covering the historical evolution of pull-through procedures (Swenson, Duhamel, Soave) through to modern laparoscopic and transanal approaches, with outcome data for each technique.
- Search PubMed — Seminal description of the modern single-stage transanal endorectal pull-through technique that has become standard of care for short-segment HD.
- Search PubMed — Prospective head-to-head comparison of rectal biopsy, anorectal manometry, and contrast enema for HD diagnosis; established the superior sensitivity and specificity of rectal biopsy as the gold standard.
- Search PubMed — Reviews the pathogenesis of HAEC including disrupted mucosal barrier function, altered microbiome, and inflammatory mediators; discusses risk factors and preventive strategies.
- Search PubMed — Orvar Swenson's personal account of developing the first curative pull-through procedure in 1948 and the clinical observations that led to understanding HD as a ganglionic disorder.
- Ieiri S et al. — Long-term outcomes and the quality of life of Hirschsprung disease in adolescents who have reached 18 years of age or older — Journal of Pediatric Surgery — PMID: 24657119 — Long-term follow-up study of HD patients into adulthood; documents fecal continence rates, QoL scores, and social outcomes; shows ongoing improvement through adolescence.
- Search PubMed — Practical clinical review of HAEC prevention, recognition, grading, and management including the role of rectal irrigations, antibiotics, and surgical intervention thresholds.
- Search PubMed — Large institutional series reporting long-term functional outcomes across multiple pull-through techniques; documents rates of constipation, soiling, HAEC recurrence, and need for redo procedures.
- Search PubMed — Classic long-term outcome series from Indiana University covering 260 patients; established benchmark mortality and functional outcome data that guided surgical decision-making for decades.
- PubMed: Hirschsprung disease RET mutation genetics — Search for current literature on RET proto-oncogene mutations and their role in HD pathogenesis, penetrance, and genetic counseling.
- PubMed: Hirschsprung disease Down syndrome epidemiology — Search for studies on the co-occurrence of HD and trisomy 21, including incidence data and management considerations in this high-risk population.
Connections
- Pediatrics
- Pyloric Stenosis
- Intussusception
- Neonatal Jaundice
- Kawasaki Disease
- Gastroenterology
- SIBO
- Constipation
- Magnesium