Galactosemia
- What Is Galactosemia?
- Biochemistry and Pathophysiology
- Newborn Screening
- Clinical Presentation (Neonatal)
- Long-Term Complications
- Diagnosis
- Treatment and Dietary Management
- Special Considerations
- Key Research Papers
- Connections
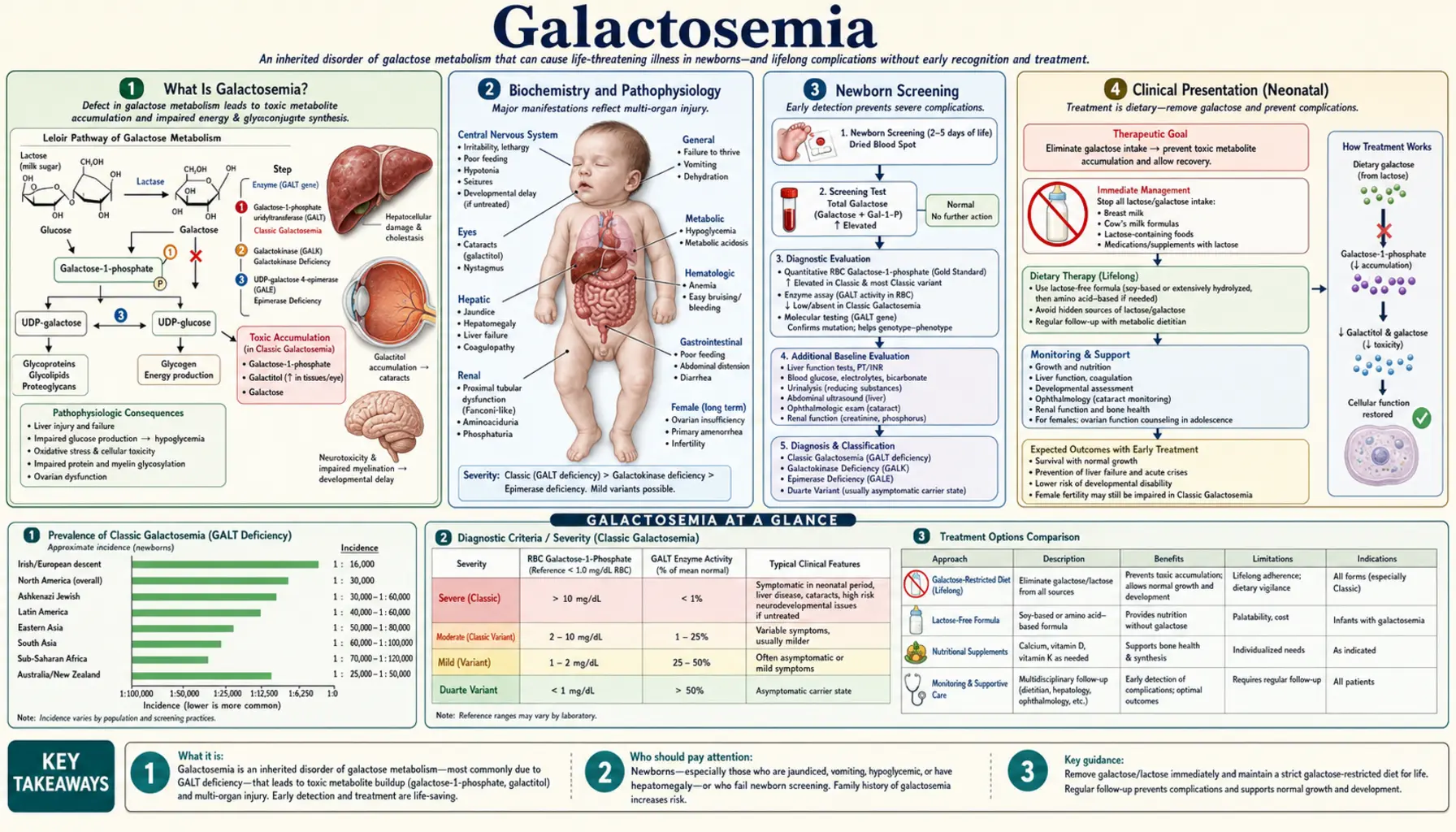
What Is Galactosemia?
Galactosemia is an inborn error of galactose metabolism — a group of inherited metabolic disorders in which the body cannot properly convert the sugar galactose into glucose. When affected infants consume breast milk or standard lactose-containing formula, galactose and its toxic metabolites accumulate rapidly in the blood, liver, brain, kidneys, and eyes, causing life-threatening organ damage within days of birth.
There are three main enzyme deficiency types, each caused by a mutation in a different gene of the Leloir pathway:
- Classic Galactosemia (Type I) — GALT gene deficiency: Deficiency of galactose-1-phosphate uridylyltransferase (GALT) is the most common and clinically severe form. Galactose-1-phosphate accumulates to toxic levels in the liver, brain, kidneys, and ovaries. Without immediate dietary intervention, neonates develop fulminant liver failure, sepsis, and death within weeks. Incidence: approximately 1 in 30,000–40,000 live births in the United States.
- Galactokinase Deficiency (Type II) — GALK gene: Deficiency of galactokinase prevents the first step of galactose phosphorylation. Galactose is shunted entirely to galactitol via aldose reductase. The primary and often only complication is bilateral cataracts from galactitol accumulation in the lens. Systemic toxicity is mild because galactose-1-phosphate does not accumulate. Incidence: approximately 1 in 150,000 births.
- Galactose Epimerase Deficiency (Type III) — GALE gene: Deficiency of UDP-galactose-4-epimerase causes a clinically variable spectrum, ranging from a benign biochemical finding confined to red blood cells (peripheral GALE deficiency — no dietary restriction needed) to a severe generalized form resembling classic galactosemia when GALE is deficient in all tissues. Incidence is rare and estimates vary widely by population.
Classic galactosemia (Type I) is the subject of most clinical guidelines, research, and newborn screening programs. It is a paradigm for how universal newborn screening can convert a formerly fatal neonatal disease into a manageable chronic condition — while also revealing the sobering reality that early dietary treatment, though lifesaving, does not prevent all long-term complications.
Biochemistry and Pathophysiology
Galactose is a monosaccharide obtained almost entirely from dietary lactose — the disaccharide in breast milk and standard infant formulas. Intestinal lactase cleaves lactose into galactose and glucose in the small intestine; galactose then enters the bloodstream and must be converted to glucose for cellular energy use. This conversion requires the Leloir pathway, a three-enzyme sequence:
- Galactokinase (GALK): Galactose + ATP → Galactose-1-phosphate + ADP
- Galactose-1-phosphate uridylyltransferase (GALT): Galactose-1-phosphate + UDP-Glucose → Glucose-1-phosphate + UDP-Galactose (this is the step blocked in classic galactosemia)
- UDP-galactose-4-epimerase (GALE): UDP-Galactose ↔ UDP-Glucose (interconversion)
In classic galactosemia (GALT deficiency), galactose-1-phosphate cannot be converted to glucose-1-phosphate. It accumulates rapidly in all cells of the body. The mechanisms of toxicity involve several interconnected pathways:
- Direct galactose-1-phosphate toxicity: Galactose-1-phosphate inhibits phosphoglucomutase (blocking glycogenolysis → hypoglycemia) and competes with glucose-1-phosphate in critical metabolic reactions. High intracellular concentrations are directly cytotoxic to hepatocytes, renal tubular cells, and neurons.
- Galactitol accumulation: Aldose reductase converts the excess galactose to galactitol, an osmotically active sugar alcohol that the body cannot metabolize further. Galactitol accumulates in the lens (causing cataracts), peripheral nerves, and brain. It is excreted in urine, providing a diagnostic marker.
- Oxidative stress: Accumulating galactose metabolites generate reactive oxygen species, contributing to mitochondrial dysfunction and cell death, particularly in the liver and brain.
- Impaired glycosylation: GALT deficiency reduces the availability of UDP-galactose, a precursor for galactosylation of glycoproteins and glycolipids. Defective glycoprotein synthesis may underlie some of the long-term neurological and ovarian complications that persist despite dietary galactose restriction.
Target organs and their vulnerabilities:
- Liver: Hepatocellular damage → jaundice, hepatomegaly, coagulopathy, cirrhosis, acute liver failure.
- Brain: Neuronal injury → intellectual disability, speech dyspraxia, ataxia, tremor; endogenous galactose synthesis (from glucose via the De Novo pathway) continues even on dietary restriction, explaining why brain damage is not fully preventable.
- Kidneys: Proximal tubular dysfunction → renal Fanconi syndrome (generalized aminoaciduria, glucosuria, phosphaturia, renal tubular acidosis).
- Ovaries: Direct galactose-1-phosphate toxicity to oocytes → germ cell depletion beginning in utero → premature ovarian insufficiency in the vast majority of females.
- Lens: Galactitol accumulation → cataracts, appearing within days to weeks of birth.
Newborn Screening
Universal newborn screening (NBS) for galactosemia is one of the great successes of preventive neonatology. All US states include galactosemia in their mandatory newborn metabolic screening panels, performed on a heel-prick blood spot at 24–48 hours of age (and repeated if the birth sample was collected before 24 hours). The primary screening test is the Beutler test — a fluorometric assay measuring GALT enzyme activity in the dried blood spot. Absent or severely reduced GALT fluorescence constitutes a positive screen.
Many states also measure total galactose (galactose + galactose-1-phosphate) on the blood spot to increase sensitivity, particularly for infants who have not yet received much oral galactose (before feeding is established) and for detecting galactokinase deficiency (Type II), which has normal GALT activity but elevated galactose. A combined enzyme-plus-metabolite approach improves sensitivity toward 100% for classic galactosemia.
A critical clinical point: Newborns with galactosemia appear entirely normal at birth. The maternal enzyme during gestation partially clears fetal galactose metabolites across the placenta. Symptoms emerge only in the days after galactose exposure from breast milk or lactose formula begins. This underscores why the 24–48 hour timing of NBS is essential — affected infants begin accumulating toxic metabolites from the first feeding. When NBS returns a positive result, galactose must be eliminated immediately, often before confirmatory testing is complete, to prevent or limit organ damage.
Infants born on weekends or holidays who receive several days of breast milk before NBS results return are at particular risk. Pediatricians and lactation consultants should be aware that a positive galactosemia screen from the state NBS laboratory is a medical emergency requiring same-day dietary change, not a result to "watch and repeat." Confirmatory diagnostic testing proceeds in parallel with dietary intervention — never in place of it.
International NBS practices vary: some countries screen universally, others do not screen at all. In regions without NBS, classic galactosemia typically presents as an acute neonatal emergency at 3–14 days of life, often diagnosed only after significant liver damage or E. coli sepsis has occurred.
Clinical Presentation (Neonatal)
Classic galactosemia presents in the first days to two weeks of life, after galactose exposure from feeding has begun. The initial picture can mimic neonatal sepsis, biliary atresia, or other causes of neonatal hepatitis — a diagnostic trap that historically delayed recognition before universal NBS. The full clinical constellation includes:
Hepatic manifestations (most prominent):
- Jaundice: Both direct (conjugated) and indirect (unconjugated) hyperbilirubinemia, reflecting both hepatocellular dysfunction and hemolysis. Jaundice typically deepens over the first week and does not respond to phototherapy if the underlying cause is not treated.
- Hepatomegaly: Liver enlargement from hepatocellular galactose-1-phosphate accumulation, fatty change, and early inflammation.
- Coagulopathy: Reduced synthesis of clotting factors (II, V, VII, X) from hepatic dysfunction. Prolonged PT and elevated INR are early warning signs of impending liver failure.
- Liver failure: Untreated infants develop progressive cirrhosis and fulminant liver failure within weeks, with ascites, portal hypertension, and hemorrhage.
Feeding and growth:
- Poor feeding, vomiting, and diarrhea — the galactose-laden feedings that cause the disease also trigger gastrointestinal intolerance.
- Failure to thrive and weight loss — inadequate caloric intake from vomiting plus metabolic demands of a failing liver.
- Hypoglycemia — hepatic gluconeogenesis and glycogenolysis are impaired by galactose-1-phosphate inhibition of phosphoglucomutase.
Cataracts: Bilateral opalescent or "oil-droplet" cataracts, visible on careful slit-lamp examination or direct ophthalmoscopy, may appear within days of birth. They result from galactitol accumulation in the lens, causing osmotic swelling and protein denaturation. Importantly, cataracts visible at this early stage may be reversible with prompt galactose elimination — a point of particular urgency.
E. coli neonatal sepsis: A sentinel and deadly complication. Galactose-1-phosphate impairs neutrophil bactericidal function, dramatically increasing susceptibility to gram-negative infection. E. coli neonatal sepsis in any newborn should be considered galactosemia until proven otherwise — this is not a teaching cliché but a life-saving clinical rule. Multiple case series have documented that the presentation of overwhelming E. coli septicemia in the first weeks of life is often the event that leads to the diagnosis of galactosemia in regions without NBS. The combination of E. coli sepsis plus jaundice plus poor feeding in a neonate demands immediate galactosemia workup and dietary galactose elimination.
Renal Fanconi syndrome: Proximal tubular dysfunction produces aminoaciduria (generalized, not specific amino acids), glucosuria (despite normal blood glucose), phosphaturia, and renal tubular acidosis. These findings on urinalysis support the diagnosis and reflect renal tubular galactose-1-phosphate toxicity.
Long-Term Complications
Classic galactosemia is unique and humbling among inborn errors of metabolism in that early and strict dietary treatment, while lifesaving, does not prevent the full spectrum of long-term complications. Families and clinicians must understand this clearly to avoid the false reassurance that a galactose-free diet equals a fully normal life. The long-term outcomes in treated classic galactosemia include:
Cognitive and intellectual impairment: Despite diet initiated in the newborn period through NBS, the majority of patients with classic galactosemia have some degree of intellectual disability or cognitive impairment. Mean IQ scores in large cohorts are typically 10–20 points below population means, with wide individual variation. Learning disabilities, particularly in mathematics and reading, are common even in patients with IQ in the normal range. The mechanism is not fully understood but is thought to involve ongoing endogenous galactose synthesis (galactose is produced from glucose via the De Novo pathway in all cells and cannot be eliminated by diet) and impaired glycosylation of brain glycoproteins and glycolipids.
Speech and language impairment: A characteristic verbal dyspraxia (childhood apraxia of speech) — difficulty with the motor planning and sequencing required for speech production — affects a large proportion of patients, often disproportionate to their general intellectual level. Expressive language is more impaired than receptive language. Speech-language therapy is a core component of management and should begin early.
Neurological manifestations: Tremor (most commonly an action or postural tremor), cerebellar ataxia, and gait abnormalities are reported in a substantial minority of adult patients. Brain MRI may show white matter abnormalities and cerebellar changes. These neurological findings tend to emerge in late childhood or adulthood and may progress despite dietary adherence.
Premature Ovarian Insufficiency (POI): The most devastating long-term complication in female patients. 80–90% of females with classic galactosemia develop premature ovarian insufficiency, defined as loss of ovarian function before age 40. The mechanism involves direct galactose-1-phosphate toxicity to oocytes — germ cell depletion begins in utero, even in fetuses whose galactosemia has not yet been recognized. By the time dietary treatment begins after birth, irreversible oocyte loss has already occurred. Clinical consequences include primary amenorrhea or secondary amenorrhea in adolescence or young adulthood, infertility, and early menopause with all its attendant risks (osteoporosis, cardiovascular risk, cognitive effects). Hormone replacement therapy is essential for health maintenance; fertility is severely compromised and spontaneous pregnancy is rare though reported.
Bone health: Calcium and vitamin D deficiency are concerns in galactosemia because the lactose-free diet eliminates the primary dietary calcium source (dairy). Without supplementation and monitoring, reduced bone mineral density and increased fracture risk are documented in adolescent and adult patients.
Psychosocial burden: The combination of cognitive difficulties, speech impairment, infertility, and lifelong dietary restriction creates substantial psychosocial challenges. Anxiety, depression, and social difficulties are reported at higher rates than in the general population. Comprehensive multidisciplinary care addressing psychological well-being is as important as metabolic monitoring.
Diagnosis
Diagnosis of classic galactosemia rests on three complementary pillars — enzyme activity measurement, metabolite levels, and molecular genetic testing — and is almost always initiated by a positive newborn screen.
GALT enzyme activity: Red blood cell GALT enzyme activity is measured in whole blood. In classic galactosemia, GALT activity is absent or near-zero (typically <1% of normal). This is the most rapid and clinically actionable confirmatory test. A result of 0–5% activity confirms diagnosis and demands immediate dietary intervention. Note: infants who have received a blood transfusion within 3 months will have artificially elevated GALT activity from donor red cells — in such cases, molecular testing is required for diagnosis.
Galactose-1-phosphate (Gal-1-P) levels: Erythrocyte galactose-1-phosphate is elevated (normal: <1 mg/dL; untreated classic galactosemia: often >10 mg/dL). Serial Gal-1-P measurements are used for ongoing dietary monitoring — the goal is to maintain erythrocyte Gal-1-P below 3–5 mg/dL, though the precise target associated with the best long-term outcomes remains debated.
Galactitol in urine: Elevated urine galactitol (measured by gas chromatography-mass spectrometry in urine organic acid analysis) provides supportive evidence and reflects the degree of galactose exposure. It normalizes rapidly with dietary restriction.
GALT gene molecular testing: DNA sequencing identifies the specific mutations in both GALT alleles. This is essential for:
- Confirming the diagnosis when enzyme activity is equivocal (e.g., after blood transfusion).
- Identifying the Duarte variant (D/G compound heterozygote): The Duarte-2 (D2) allele (p.N314D) reduces GALT enzyme activity to approximately 25% of normal when compound-heterozygous with a classic galactosemia mutation. Duarte galactosemia is generally mild and may not require dietary restriction — clinical outcomes data are reassuring, though some centers still advise a time-limited dietary restriction in infancy while evidence accrues.
- The most common pathogenic variant in Caucasian populations is p.Q188R, associated with near-complete GALT deficiency and the most severe phenotype. The p.S135L variant is more common in African-American patients and tends to be associated with a somewhat milder biochemical but not necessarily milder clinical phenotype.
- Carrier testing for family members and prenatal diagnosis in subsequent pregnancies.
Ophthalmologic evaluation: Slit-lamp examination for cataracts should be performed at diagnosis and at regular intervals. Early cataracts (from galactitol) regress with dietary restriction; late-onset cataracts (from galactose-1-phosphate toxicity) may be irreversible.
Newborn sibling testing: Any sibling of a child with classic galactosemia should be tested immediately at birth — by cord blood enzyme assay or by urgent blood spot — before dietary galactose exposure begins. Waiting for routine NBS results is acceptable only if results are available within 24 hours; otherwise, presumptive dietary restriction pending results is appropriate given the risk.
Treatment and Dietary Management
Immediate dietary galactose elimination is the cornerstone of treatment and must be implemented at once upon a positive newborn screen — before confirmatory testing is complete. This means stopping breast milk and lactose-containing formula immediately and switching to a galactose-free formula:
- Soy-based infant formula (e.g., Similac Soy Isomil, Enfamil ProSobee): Soy protein contains no lactose. Note: soy oligosaccharides (stachyose, raffinose) are metabolized to some galactose in the gut — this was historically a concern, but clinical evidence shows soy formula is clinically safe and nutritionally adequate for galactosemia.
- Casein hydrolysate formula (e.g., Nutramigen, Alimentum): Lactose-free; useful for infants who cannot tolerate soy protein (allergy, preference).
Lifelong galactose restriction remains the standard of care, though the strictness and evidence base continue to evolve. Key dietary principles:
- Eliminate dairy products (milk, cheese, yogurt, butter, cream, ice cream) — the primary galactose source.
- Check medications for lactose excipient: Many tablets and capsules use lactose as a filler. Patients and pharmacists must review each medication; lactose-free alternatives or liquid formulations are used when needed.
- Certain vegetables and legumes contain small amounts of galactose (beets, peas, lentils, chickpeas, soybeans) — recommendations vary by center regarding strict avoidance vs. modest intake. Clinical practice is heterogeneous on this point.
- Organ meats (liver, kidney, brain) — high in galactosyl-containing glycolipids; many centers advise avoidance.
- Fermented dairy (yogurt, aged cheese): Bacterial fermentation degrades much of the lactose; some guidelines permit moderate fermented dairy in older patients, particularly given the endogenous galactose synthesis that continues regardless of dietary intake. This remains a subject of active clinical debate.
Calcium and vitamin D supplementation is essential. Eliminating dairy from birth creates a calcium deficit that, if uncorrected, leads to impaired bone mineralization. Supplemental elemental calcium (500–1000 mg/day depending on age) plus vitamin D3 (600–1000 IU/day) should be prescribed from infancy. Bone density (DEXA scan) should be monitored from adolescence.
Monitoring erythrocyte galactose-1-phosphate: Regular Gal-1-P measurement (every 3–6 months in infancy, annually in stable older patients) guides dietary compliance and identifies galactose exposure from hidden sources. Elevated levels despite apparent dietary adherence should prompt a detailed dietary review.
Multidisciplinary care team: Optimal management requires a metabolic dietitian (dietary galactose management, calcium, overall nutrition), a metabolic biochemist or metabolic pediatrician (biochemical monitoring), a developmental pediatrician or neuropsychologist (cognitive and speech follow-up), a speech-language pathologist (verbal dyspraxia treatment), an ophthalmologist (cataracts), and a reproductive endocrinologist (POI management in females from adolescence onward).
Special Considerations
Premature Ovarian Insufficiency (POI) — Hormone Replacement Therapy: The majority of females with classic galactosemia will develop POI, typically manifesting as primary or secondary amenorrhea in adolescence. When FSH is elevated and estradiol is low, hormone replacement therapy (HRT) with estrogen and progesterone is indicated — both to manage menopausal symptoms and to protect bone density and cardiovascular health. HRT does not restore fertility. It should be initiated promptly when POI is diagnosed, not delayed, as the window for bone mineral accrual in adolescence is narrow.
Fertility preservation: Because POI is expected but not universal, and because some patients retain partial ovarian function for a period before full POI develops, oocyte or ovarian tissue cryopreservation before complete POI is an option for post-pubertal females. Counseling about fertility and POI should begin in early adolescence. The decision to pursue fertility preservation is personal and complex; a reproductive endocrinologist experienced with POI should be involved.
Duarte galactosemia (D/G compound heterozygotes): The Duarte-2 allele reduces GALT activity to roughly 25% of normal when compound-heterozygous with a classic galactosemia mutation. Population-based cohort studies (e.g., Waisbren et al.) have not demonstrated significant developmental, cognitive, or speech-language differences between Duarte galactosemia and unaffected controls when dietary restriction is not used. Most expert centers in the US no longer recommend dietary galactose restriction for Duarte galactosemia; others still advise time-limited restriction during infancy while monitoring Gal-1-P levels. Families should receive transparent counseling about the evidence and its limitations.
Galactosemia in pregnancy: Females with classic galactosemia who achieve spontaneous pregnancy (rare but documented) should be advised of theoretical risks of endogenous fetal galactose exposure; strict maternal galactose restriction is generally continued. Newborn offspring should be tested immediately at birth. There is also a parallel with maternal PKU: just as untreated maternal phenylketonuria harms the fetus even when the fetus does not have PKU, it is theoretically possible that maternal galactosemia increases fetal galactose exposure, though the magnitude of this effect is less well characterized than in maternal PKU because POI makes such pregnancies rare.
Emerging therapies: Research into therapies beyond dietary restriction is active. Investigational approaches include: enzyme replacement therapy (challenges include intracellular enzyme delivery); substrate reduction strategies; galactose-1-phosphate sequestration; and mRNA/gene therapy approaches. None are yet in clinical use. The development of a robust biomarker that predicts long-term outcome independently of Gal-1-P levels is a key research priority, as current monitoring tools do not reliably predict cognitive trajectory or ovarian reserve.
Newborn sibling management: When a sibling of a known galactosemia patient is born, the pediatrician and delivery hospital should be notified in advance. Cord blood enzyme assay and immediate galactose-free feeding pending results is the protocol in most expert centers. Reactive NBS results — waiting until day 2–3 — risk galactose exposure during those initial days.
Key Research Papers
- Search PubMed PMID: 16601863
- Search PubMed PMID: 2124093
- Fridovich-Keil JL et al. Galactosemia. GeneReviews. 2006 (updated periodically). — PubMed search
- Search PubMed PMID: 11264421
- Search PubMed PMID: 10654876
- Search PubMed PMID: 18578876
- Search PubMed PMID: 904955
- Robertson A et al. Galactosemia: diagnosis and management. Paediatr Child Health. 2011. — PubMed search
- Search PubMed PMID: 16378706
- Search PubMed PMID: 27826746
- Search PubMed PMID: 19945932
- Search PubMed PMID: 8440781
Connections
- Pediatrics
- Congenital Heart Disease
- Neonatal Jaundice
- Endocrinology
- Lab Tests
- Vitamins
- Minerals
- Gastroenterology

Osmosis — Galactosemia pathophysiology and clinical features explained.

Ninja Nerd — Inborn errors of galactose and fructose metabolism.

Pediatric EM Morsels — Newborn metabolic screening panel and galactosemia detection.

Biochemistry Ninja — The Leloir pathway: galactose metabolism step by step.

Lecturio Medical — Neonatal jaundice: differentiating metabolic causes from physiologic.

NORD Rare Diseases — Premature ovarian insufficiency in classic galactosemia.

Pediatric Pearls — E. coli neonatal sepsis: the galactosemia connection every clinician must know.

Metabolic Support UK — Practical galactose-free diet management for galactosemia families.

ACMG Foundation — Long-term neurological and cognitive outcomes in classic galactosemia.

Boards and Beyond — USMLE review: inborn errors of metabolism in pediatrics.

American Academy of Ophthalmology — Recognizing early galactosemia cataracts in the newborn eye exam.

Dietitian Connection — Calcium and vitamin D supplementation strategies for dairy-free metabolic diets.

University Metabolic Clinic — Duarte galactosemia: distinguishing benign variant from classic disease.

Baby's First Test — How newborn heel-prick screening detects galactosemia and metabolic disorders.

ASHA — Verbal dyspraxia in galactosemia: speech-language therapy approaches for affected children.

Resolve Fertility — Oocyte cryopreservation and fertility options for women with premature ovarian insufficiency.