Congenital Hypothyroidism
- What Is Congenital Hypothyroidism?
- Normal Thyroid Development and Function
- Newborn Screening
- Clinical Presentation
- Cretinism
- Diagnosis
- Treatment
- Permanent vs. Transient Hypothyroidism
- Neurodevelopmental Outcomes
- Key Research Papers
- Connections
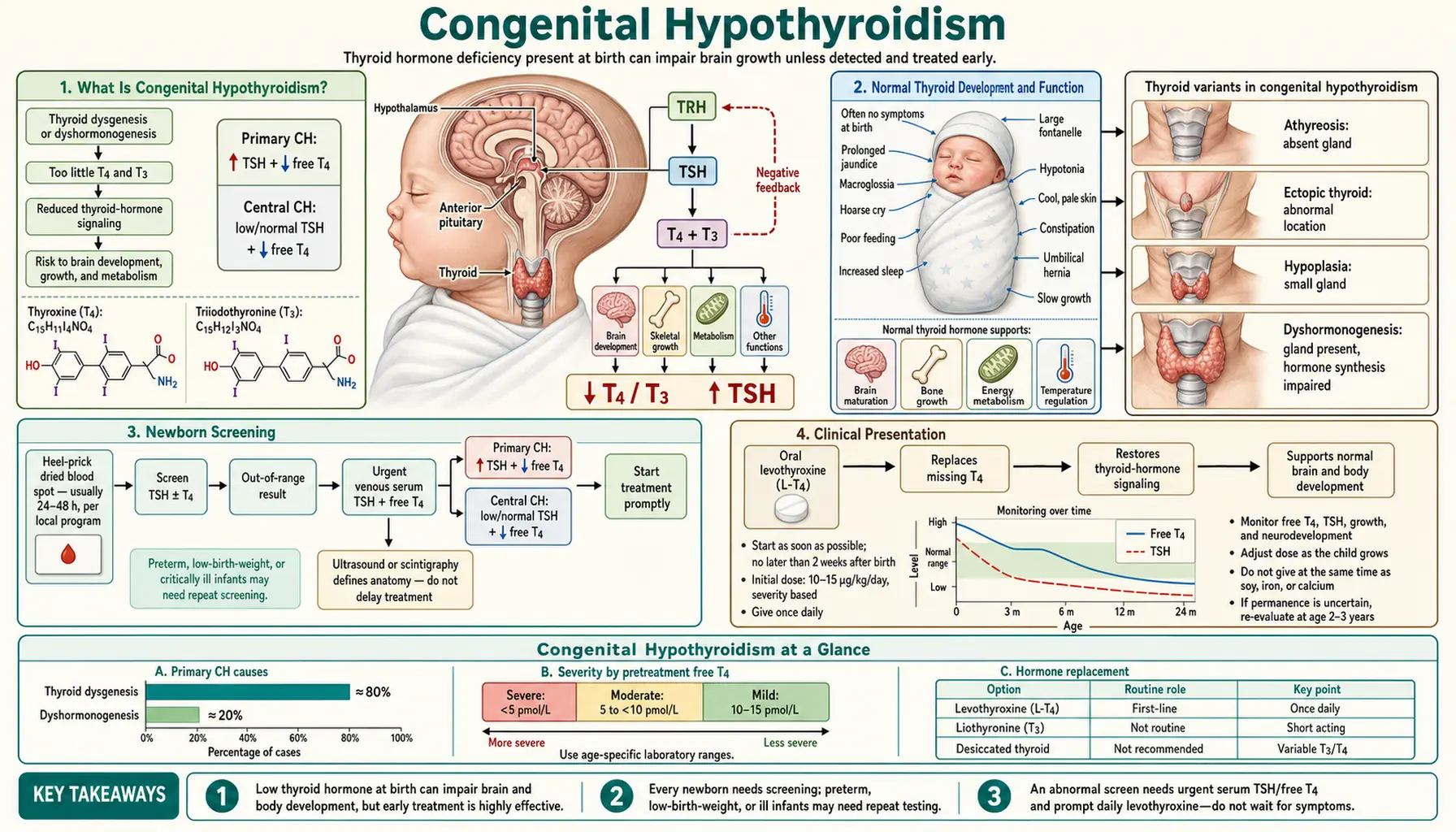
What Is Congenital Hypothyroidism?
Congenital hypothyroidism (CH) is a condition in which the thyroid gland is absent, underdeveloped, or dysfunctional from birth, resulting in insufficient thyroid hormone production from the very start of life. It is the most common preventable cause of intellectual disability worldwide and the most common congenital endocrine disorder, affecting approximately 1 in 2,000 to 3,000 newborns in iodine-sufficient countries. In the United States, this translates to roughly 1,500–2,000 affected infants each year.
Because thyroid hormone is essential for brain maturation — particularly the myelination of nerve fibers, neuronal migration, and synapse formation — untreated CH causes permanent, irreversible intellectual disability and growth failure. The profound tragedy of this disorder is that, with early diagnosis and treatment, the vast majority of affected children develop completely normally. Universal newborn screening has transformed CH from a leading cause of intellectual disability into a highly manageable condition.
Etiology: Four Main Types
1. Thyroid Dysgenesis (85% of cases) — abnormal development or migration of the thyroid gland during fetal life. Subtypes include:
- Agenesis/athyreosis: Complete absence of thyroid tissue — the most severe form, causing permanent hypothyroidism requiring lifelong treatment.
- Ectopic thyroid: Thyroid tissue present but located in the wrong place along the thyroglossal duct migration path. The sublingual (base of tongue) position is the most common ectopic site. Ectopic tissue is often insufficient to meet the body's hormonal demands.
- Hypoplastic thyroid: A small, structurally present but functionally inadequate gland in the normal position.
2. Dyshormonogenesis (15% of cases) — the thyroid gland is structurally present and normally positioned, but enzyme defects impair the synthesis of thyroid hormone. Mutations in TPO (thyroid peroxidase), TG (thyroglobulin), and DUOX2 (dual oxidase 2) genes are the most commonly identified causes. These defects are transmitted in an autosomal recessive pattern and often produce a goiter (enlarged thyroid), because the gland compensates by growing larger in response to TSH stimulation. Dyshormonogenesis should be suspected when CH occurs in a sibling, pointing to the hereditary nature of the defect.
3. Central (Secondary/Tertiary) Hypothyroidism (rare) — caused not by a thyroid gland problem but by deficiency of thyroid-stimulating hormone (TSH) from the pituitary gland (secondary) or thyrotropin-releasing hormone (TRH) from the hypothalamus (tertiary). Because TSH-based newborn screening programs will miss this type (TSH is low or normal rather than elevated), affected infants may escape detection unless T4-based screening is used. Central CH is often associated with other pituitary hormone deficiencies (growth hormone, cortisol) and structural midline brain abnormalities.
4. Transient Causes — hypothyroidism that resolves spontaneously, usually within months. Key causes include:
- Maternal antithyroid drug use: Propylthiouracil (PTU) or methimazole used to treat maternal Graves' disease cross the placenta and suppress the fetal thyroid.
- Iodine deficiency or excess: Iodine deficiency is the world's leading preventable cause of intellectual disability and impairs thyroid hormone synthesis. Paradoxically, iodine excess (from iodine-containing antiseptics used on premature infants or in neonatal care) can suppress the fetal thyroid via the Wolff-Chaikoff effect.
- Maternal TSH-receptor blocking antibodies: In women with autoimmune thyroid disease (Hashimoto's thyroiditis, prior Graves' disease), blocking antibodies against the TSH receptor can cross the placenta and suppress the fetal thyroid gland. These maternal antibodies clear from the infant's circulation over weeks to months, at which point thyroid function normalizes.
Normal Thyroid Development and Function
The human thyroid gland develops from the thyroglossal duct, a midline outgrowth from the floor of the primitive pharynx (foramen cecum, at the junction of the anterior two-thirds and posterior one-third of the tongue). Beginning around gestational week 3–4, the thyroid anlage migrates caudally, passing anterior to the hyoid bone and trachea, to reach its final position in the anterior neck by week 7. The thyroglossal duct normally involutes and disappears; its persistence leads to a thyroglossal duct cyst. Failure of normal migration produces the ectopic thyroid locations seen in thyroid dysgenesis.
The thyroid gland synthesizes two hormones: thyroxine (T4, tetraiodothyronine) — the major secretory product — and triiodothyronine (T3), the biologically active form. Synthesis requires adequate dietary iodine: iodide is actively trapped by the sodium-iodide symporter (NIS), oxidized by thyroid peroxidase (TPO), and incorporated into the tyrosine residues of thyroglobulin (TG) to form monoiodotyrosine (MIT) and diiodotyrosine (DIT). Coupling of DIT + DIT forms T4; DIT + MIT forms T3. The stored thyroglobulin-hormone complex is released into the circulation when needed.
Most T3 in the body is generated not in the thyroid but by peripheral deiodination of T4 — removal of one iodine atom — by deiodinase enzymes in target tissues including the brain, liver, and kidney. T3 is 3–5 times more potent than T4 and acts by binding to nuclear thyroid hormone receptors (TR-alpha, TR-beta) to regulate gene transcription.
Thyroid hormone and brain development are inseparable. T3 is critical for:
- Myelination: The fatty insulation of nerve axons that enables rapid signal transmission. Hypothyroidism delays and impairs myelination, producing the cognitive and motor deficits of untreated CH.
- Neuronal migration: The movement of newly born neurons from the ventricular zone to their final cortical positions, a process highly sensitive to thyroid hormone.
- Synaptogenesis: Formation of synaptic connections between neurons.
- Cerebellar development: The cerebellum, which coordinates movement and posture, is particularly thyroid-hormone-dependent in the postnatal period.
During the first trimester of pregnancy, before the fetal thyroid becomes functional (around week 12), the fetus depends entirely on maternal T4 crossing the placenta. This maternal contribution continues to be significant throughout gestation, even after the fetal thyroid begins producing its own hormone. Maternal hypothyroidism — even subclinical — during the first trimester has been associated with subtle neurodevelopmental effects in offspring, which is why thyroid function testing and iodine supplementation during pregnancy are public health priorities.
Newborn Screening
Universal newborn screening (NBS) for congenital hypothyroidism is one of the great success stories of preventive medicine. Introduced in North America in the early 1970s following pioneering work by Jean Dussault in Quebec, it is now mandated in all 50 US states, across the European Union, Australia, Japan, and many other countries.
The heel prick: A few drops of blood are collected from the newborn's heel onto a filter paper card at 24–48 hours of age (ideally after 24 hours, when TSH levels have normalized from the physiological TSH surge at birth). The dried blood spot is sent to a state laboratory for analysis.
Two screening strategies are used:
- Primary TSH screening (most common in Europe and much of the world): Measures TSH alone. An elevated TSH signals primary hypothyroidism (thyroid gland failure). Simple, sensitive, and specific for the most common forms of CH. Will miss central (secondary/tertiary) hypothyroidism where TSH is inappropriately low.
- Primary T4 + backup TSH (used in some US states): Measures T4 first; a low T4 triggers TSH measurement. Detects both primary CH (high TSH) and central CH (low TSH). Slightly higher false-positive rate than TSH-primary screening.
The critical clinical insight is that most newborns with CH look completely normal at birth. This is because maternal T4 crosses the placenta throughout pregnancy, providing enough thyroid hormone to prevent obvious neurological damage in utero and to suppress obvious clinical signs in the first days of life. As the maternal T4 is gradually cleared from the infant's circulation over the first weeks after birth, clinical signs of hypothyroidism emerge — but by then, the diagnosis would be devastatingly delayed without screening. This is why NBS — not clinical recognition — is the cornerstone of diagnosis.
A positive screening result (elevated TSH or low T4) requires urgent confirmatory testing with serum TSH and free T4, followed by imaging. Treatment should begin within 2 weeks of birth to optimize neurodevelopmental outcomes.
Clinical Presentation
Without newborn screening — or when diagnosis is delayed — clinical signs of congenital hypothyroidism emerge gradually over the first weeks to months of life as maternal thyroid hormone clears. The constellation of findings reflects the systemic effects of thyroid hormone deficiency on growth, metabolism, and brain development.
Classic Signs and Symptoms
- Prolonged jaundice (neonatal hyperbilirubinemia): One of the earliest and most common signs. Thyroid hormone is required for normal maturation of bilirubin conjugation enzymes. Hypothyroid infants have prolonged physiological jaundice that lasts beyond 2 weeks (the point at which jaundice in a term neonate is considered pathological until proven otherwise). Clinicians should test thyroid function in any infant with unexplained prolonged jaundice.
- Large posterior fontanelle: The posterior fontanelle normally closes by 6–8 weeks of age; delayed closure (or an unusually large posterior fontanelle at birth, >0.5 cm) reflects impaired ossification due to thyroid hormone deficiency.
- Macroglossia (large tongue): The tongue may protrude from the mouth, interfering with feeding. Results from myxedematous deposits in the tongue tissue.
- Constipation: Slowed intestinal motility from reduced metabolic rate. May be severe.
- Hypotonia (floppy infant): Decreased muscle tone is a consistent finding, contributing to feeding difficulties, poor suck, and delayed motor milestones.
- Umbilical hernia: Weakness of the abdominal wall musculature, combined with abdominal distension from constipation and hypotonia, produces protrusion at the umbilicus.
- Hoarse cry: Myxedematous changes in the larynx and vocal cords produce a characteristically low-pitched, coarse cry.
- Dry, mottled skin: Reduced sweat gland activity and myxedematous skin infiltration cause cool, dry, pale or mottled skin.
- Puffy face (myxedema): Accumulation of glycosaminoglycans in the dermis produces non-pitting facial and periorbital puffiness.
- Goiter: Present in dyshormonogenesis (where the defect is in hormone synthesis, not gland formation), as TSH drives compensatory thyroid gland enlargement. Absent in thyroid dysgenesis.
- Bradycardia and hypothermia: Reduced metabolic rate lowers heart rate and body temperature.
- Feeding difficulties: Poor suck reflex, lethargy, and macroglossia all impair breastfeeding. Affected infants may be sleepy and difficult to rouse for feeds.
If hypothyroidism remains untreated through the first months and years of life, the devastating consequence is permanent intellectual disability — the clinical picture historically known as cretinism. This outcome is entirely preventable with early diagnosis and treatment.
Cretinism
Cretinism is the syndrome of severe intellectual disability, growth retardation, deafness, and neurological impairment resulting from profound, untreated thyroid hormone deficiency from early in life. The word "cretin" derives from the French Savoyard dialect term crestin ("Christian"), a reminder that those so affected were still fully human despite their disability — a humanizing designation in its historical context, though the term is now considered offensive in everyday language and has been replaced in medical literature by "congenital hypothyroidism with intellectual disability."
Neurological Cretinism
The most severe form, caused by profound iodine deficiency during fetal development, is called neurological cretinism. Because the fetus depends on maternal T4 during the critical period of brain development (first and early second trimester), severe iodine deficiency in the mother deprives the fetal brain of thyroid hormone at the most vulnerable developmental window. Features include:
- Severe intellectual disability (IQ often below 20)
- Spastic diplegia (stiff legs, difficulty walking)
- Deaf-mutism (bilateral sensorineural hearing loss)
- Squinting (strabismus)
- Normal thyroid size in the child (the iodine deficiency damaged the fetal brain; the child's thyroid may later compensate)
Myxedematous Cretinism
Caused by severe neonatal and postnatal iodine deficiency combined with thyroid gland atrophy or dysfunction, features include:
- Severe intellectual disability
- Short stature (dwarfism from growth hormone axis disruption)
- Myxedema (skin and facial puffiness)
- Hypothyroid features (bradycardia, constipation, hypotonia)
- Less severe neurological deficits than neurological cretinism
Endemic Cretinism
In iodine-depleted mountain regions — historically the Alps, Himalayas, Andes, and inland Africa — endemic cretinism was once a major public health crisis affecting entire communities. Iodization of salt, introduced broadly in the early 20th century and now implemented in most countries, has largely eliminated endemic cretinism where programs are maintained. The World Health Organization estimates that iodine deficiency remains the world's leading preventable cause of intellectual disability, affecting an estimated 50 million people in iodine-insufficient regions.
In countries with universal newborn screening and adequate iodine nutrition, cretinism is now rare. The challenge today lies in nations where NBS has not yet been fully implemented and in populations with iodine deficiency — underscoring that these public health investments save not just lives but the full cognitive potential of entire generations.
Diagnosis
Diagnosis of congenital hypothyroidism is a sequential process moving from screening to confirmation to anatomical characterization.
Step 1: Newborn Screening Recall
An abnormal NBS result (elevated TSH on filter paper) prompts urgent outpatient or inpatient evaluation. Time matters: every day of delay in treatment is associated with measurable neurodevelopmental risk.
Step 2: Confirmatory Serum Testing
Confirmatory laboratory testing includes:
- Serum TSH: Elevated in primary hypothyroidism (thyroid gland failure). The degree of elevation correlates roughly with severity — infants with TSH >100 mIU/L have severe deficiency.
- Free T4 (fT4): Low in overt hypothyroidism. The absolute fT4 level at presentation helps guide urgency and initial dosing.
- Total T4: Some labs measure total T4; interpretation requires knowledge of thyroid-binding globulin levels, which vary in the neonatal period.
- Thyroglobulin (Tg): A marker of functioning thyroid tissue. Detectable or high Tg suggests some thyroid tissue is present (ectopic or hypoplastic); undetectable Tg strongly suggests agenesis/athyreosis.
Step 3: Thyroid Imaging
Thyroid ultrasound is the first-line imaging tool. It identifies the presence, size, and location of thyroid tissue in the neck without radiation. It can distinguish a normally positioned but small gland (hypoplasia) from absent tissue. However, ultrasound cannot detect ectopic thyroid tissue located above the neck (sublingual position).
Radionuclide thyroid scan (scintigraphy) uses either technetium-99m pertechnetate (Tc-99m) — which is trapped by the thyroid like iodine but not organified — or iodine-123 (I-123). Scintigraphy visualizes the location and functional activity of all thyroid tissue, including ectopic sublingual tissue that ultrasound misses. It can distinguish athyreosis (no uptake anywhere) from ectopic thyroid (uptake at an unusual location) from dyshormonogenesis (normal or increased uptake in a normally positioned gland that is biochemically dysfunctional). Because treatment should begin immediately, imaging should not delay the start of levothyroxine; it can be performed after the first dose if necessary.
Associated Investigations
- Karyotype or chromosomal microarray: Down syndrome (trisomy 21) has an approximately 1 in 50 risk of CH — nearly 60 times the general population rate. Dysmorphic features should prompt chromosomal analysis.
- Maternal thyroid antibodies (TSH-receptor blocking antibodies): If transient CH is suspected, maternal anti-TSHR blocking antibodies can be measured; their presence explains transient suppression of the infant's thyroid.
- Genetic testing: In suspected dyshormonogenesis (family history, goiter, thyroid tissue present on imaging), genetic panel testing for TPO, TG, DUOX2, SLC5A5 (NIS), SLC26A4 (pendrin), and TSHR mutations is available at specialized centers.
Treatment
Treatment of congenital hypothyroidism is straightforward in principle and profoundly consequential in its timing: oral levothyroxine (LT4), started as early as possible, ideally within the first 2 weeks of life. Studies consistently show that the window of treatment initiation is one of the strongest predictors of IQ outcome — delays of even 1–2 weeks in severe cases are associated with measurable decrements in cognitive performance.
Levothyroxine (LT4)
Levothyroxine is synthetic T4 — chemically identical to the T4 produced by the human thyroid gland. It is the treatment of choice, not T3, because:
- T4 has a longer half-life (~7 days vs. T3's ~1 day), providing stable hormone levels.
- T4 crosses the blood-brain barrier and is locally converted to T3 in the brain by deiodinases — allowing the brain to self-regulate its T3 supply.
- T3 supplementation produces supraphysiological T3 peaks followed by troughs, which is potentially harmful to the developing brain.
Starting dose: 10–15 micrograms per kilogram per day (mcg/kg/day). This is a significantly higher weight-based dose than used in adult hypothyroidism (1.6 mcg/kg/day) because infants have much higher metabolic rates and T4 turnover. Neonates with severe hypothyroidism (TSH >100, fT4 undetectable) are started at the higher end of this range or even at 15 mcg/kg to rapidly normalize thyroid function.
Formulation and administration: Levothyroxine for infants is given as a crushed tablet mixed with a small amount (1–2 mL) of water or breast milk. Key administration cautions:
- Do NOT mix with soy-based formula: Soy phytates significantly reduce LT4 absorption, potentially by 30–50%. Soy infant formula should be avoided or, if necessary, the dose adjusted upward and monitored closely.
- Do NOT give with iron supplements or calcium: Both significantly reduce LT4 absorption. Space LT4 at least 4 hours from iron or calcium-containing supplements or fortified foods.
- Give at the same time each day, preferably in the morning, separated from feeds by 30 minutes if possible.
- If a dose is missed, give the missed dose the same day — do not skip.
Monitoring
Close monitoring of thyroid function is essential throughout infancy and childhood because the brain's requirement for thyroid hormone is highest in the first 2–3 years of life:
- First year: Serum TSH and fT4 every 1–3 months.
- Second and third years of life: Every 2–3 months.
- After age 3: Every 3–12 months depending on stability.
Treatment targets: The goal is to maintain serum TSH in the low-normal range (approximately 0.5–2 mIU/L in most guidelines) and free T4 in the upper half of the normal range for age. Over-treatment (suppressed TSH) is associated with accelerated bone maturation and craniosynostosis in infants; under-treatment (elevated TSH) risks ongoing neurological harm.
Dose adjustments: Because infants grow rapidly, the absolute levothyroxine dose must be adjusted frequently — at nearly every monitoring visit in the first year. Parents should be counseled that dose changes are routine and expected, not signs of treatment failure.
Permanent vs. Transient Hypothyroidism
A clinically important question in every case of congenital hypothyroidism is whether the condition is permanent (lifelong treatment required) or transient (thyroid function will normalize spontaneously). Approximately 20–30% of CH cases identified by newborn screening are transient, particularly in premature infants and those born to mothers with autoimmune thyroid disease or who received iodine-containing antiseptics in the NICU.
Predicting Transient vs. Permanent
Features suggesting permanent CH:
- Thyroid agenesis or severe ectopia on imaging
- Very high TSH (>100 mIU/L) and very low fT4 at diagnosis
- Dyshormonogenesis (mutation identified in thyroid hormone synthesis genes)
Features suggesting transient CH:
- Normally positioned gland of normal size on imaging
- Known maternal antithyroid drug use or maternal TSH-receptor blocking antibodies
- History of NICU iodine exposure
- Mild TSH elevation with near-normal fT4
- Premature birth
The Trial Off Levothyroxine at Age 3
Current guidelines recommend that most children with CH undergo a trial off levothyroxine around age 3 years to determine whether hypothyroidism is permanent. This age is chosen because:
- The brain's most sensitive period for thyroid hormone dependence is largely complete by age 3.
- A brief period (30 days) without treatment is unlikely to cause harm in a 3-year-old if the child has underlying thyroid function.
Protocol: Levothyroxine is discontinued for 30 days. Serum TSH is then measured. If TSH remains in the normal range, the CH was transient and treatment can be permanently discontinued. If TSH is elevated, permanent hypothyroidism is confirmed and lifelong LT4 is resumed.
An alternative approach is to reduce the LT4 dose by 30–50% at age 3 and recheck TSH after 4 weeks; a rising TSH on the reduced dose confirms permanent CH without a full 30-day off-treatment period.
Genetic testing for dyshormonogenesis is increasingly offered in the workup of CH, particularly when imaging shows a normally positioned gland (making athyreosis/ectopia less likely). Identification of biallelic mutations in TPO, DUOX2, or other synthesis genes confirms permanent CH and makes the trial off unnecessary.
Neurodevelopmental Outcomes
The transformation wrought by universal newborn screening on the intellectual outcomes of congenital hypothyroidism is one of the most compelling demonstrations of the power of preventive medicine. In the pre-screening era, untreated CH was the leading single identifiable cause of intellectual disability, with average IQs of 30–60 in severely affected individuals. Today, with early treatment, the majority of children with CH achieve IQ scores within the normal population range.
IQ Outcomes with Early Treatment
Multiple long-term cohort studies following children diagnosed and treated through NBS have established that treatment beginning before 2 weeks of age — particularly with high initial levothyroxine doses — produces average IQ scores in the normal range (mean IQ 95–105 in most series). The Rovet (2005) review of long-term cognitive outcomes found that children with mild CH identified through NBS are virtually indistinguishable from their unaffected siblings. Even children with severe athyreosis treated very early achieve normal IQ in most studies.
Subtle Persistent Deficits
Despite normal IQ in most children with early-treated CH, subtle neurocognitive differences are consistently found in population-level studies, even with optimal treatment:
- Processing speed: Slightly slower information processing compared to matched controls.
- Verbal memory: Minor deficits in verbal learning and recall.
- Attention: Increased rates of attention difficulties, though not reaching ADHD diagnostic criteria in most studies.
- Motor coordination: Mild fine-motor and gross-motor delays, likely related to cerebellar vulnerability during the postnatal period.
These subtle differences are most pronounced in children who had the most severe initial biochemistry (lowest fT4 at diagnosis) and those with athyreosis — reflecting the biological impact of the period between birth and treatment initiation, however brief. This underscores why the goal is not just early treatment but the earliest possible treatment.
School Performance and Long-Term Follow-Up
Children with CH should receive routine developmental surveillance at pediatric well-child visits. Those with severe initial hypothyroidism benefit from:
- Early intervention programs (birth to age 3): Physical therapy, occupational therapy, and speech-language pathology referral if any developmental delays are noted.
- Individualized education plans (IEPs) if school-age academic difficulties emerge.
- Neuropsychological testing if parents or teachers identify learning difficulties, to characterize specific areas of strength and weakness and guide educational support.
The overarching message for families is one of tremendous hope: with early diagnosis through newborn screening and consistent levothyroxine treatment with regular monitoring, most children with congenital hypothyroidism live completely normal lives with normal intelligence. The condition is one of the genuine success stories of 20th-century pediatric medicine — a once-devastating disorder transformed into a highly manageable, treatable condition through public health innovation and basic science.
Key Research Papers
- Search PubMed PMID: 21396582
- Search PubMed PMID: 20920353
- Search PubMed PMID: 16785390
- Search PubMed PMID: 8419068
- Search PubMed PMID: 10969259
- Search PubMed PMID: 16231951
- Search PubMed PMID: 17356049
- Search PubMed PMID: 16033918
- Search PubMed PMID: 25257907
- Search PubMed PMID: 15897280
- Foley TP Jr. Congenital hypothyroidism. Neonatal Screening for Inborn Errors of Metabolism. 1985. (PubMed search — historical reference)
- Olivieri A et al. Epidemiological study on the prevalence of congenital hypothyroidism. Eur J Endocrinol. 2002. (PubMed search — epidemiology)
Connections
- Pediatrics
- Endocrinology
- Congenital Heart Disease
- Neonatal Jaundice
- Lab Tests
- Vitamin D — Thyroid and Immune Connections
- Iodine — Thyroid Hormone Synthesis
- Hashimoto's Thyroiditis
- Hypothyroidism — the general underactive-thyroid condition, its adult presentation and levothyroxine management.















