Merkel Cell Carcinoma
- What is Merkel Cell Carcinoma?
- Etiology: Merkel Cell Polyomavirus (MCPyV) vs UV-Induced
- Immunosuppression and Risk Factors
- Clinical Presentation
- Diagnosis: Pathology, Immunohistochemistry, and Staging
- Treatment: Surgery, Radiation, and Sentinel Lymph Node Biopsy
- Immunotherapy: Avelumab, Pembrolizumab, and the Revolution in MCC
- Prognosis, Surveillance, and MCPyV Serology as Biomarker
- Special Populations: Immunocompromised Patients
- Key Research Papers
- Connections
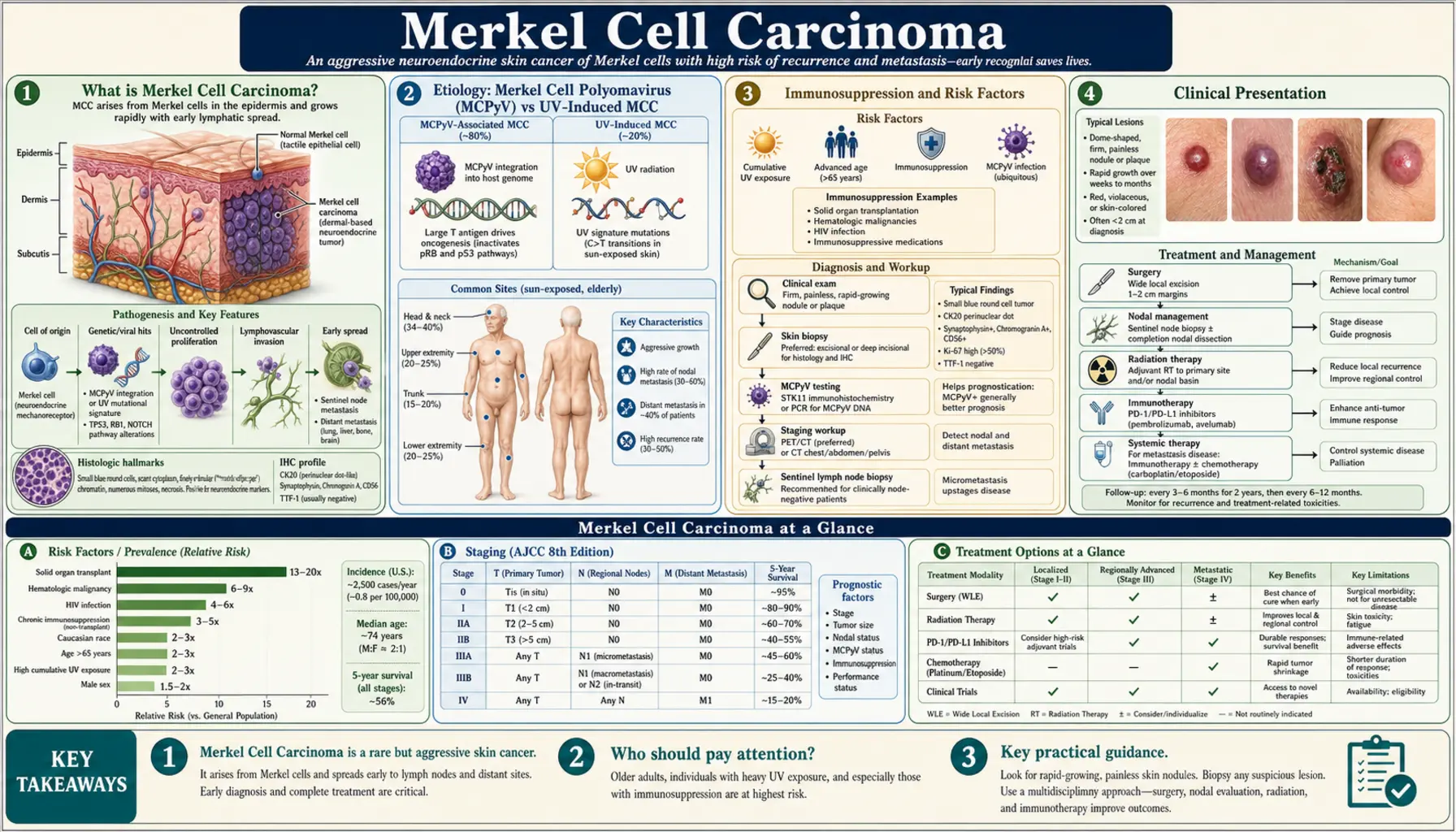
What is Merkel Cell Carcinoma?
Merkel cell carcinoma (MCC) is a rare but highly aggressive neuroendocrine malignancy of the skin. It arises from Merkel cells — specialized mechanoreceptor cells located in the basal layer of the epidermis that play a role in light-touch sensation. These cells are thought to derive from either neural crest precursors or epidermal progenitors, and their malignant transformation produces a tumor with a distinctly high metastatic potential and substantial mortality.
Approximately 3,000 new cases are diagnosed in the United States each year, a number that has been rising at roughly 8% annually over recent decades — an increase attributed in part to an aging population, rising rates of immunosuppression (from organ transplantation and certain cancers), and greater clinical recognition. Globally, MCC remains rare but is similarly trending upward.
MCC carries a worse prognosis than melanoma at equivalent stages. The five-year survival rate for localized disease is approximately 75%, dropping to 45% with regional lymph node involvement and to around 20% for distant metastatic disease. Historically, treatment outcomes were poor, but the advent of immunotherapy beginning in 2016–2018 transformed the management landscape for advanced MCC, producing durable responses in a meaningful fraction of patients.
The disease was first formally described in 1972 by pathologist Cyril Toker, who called it "trabecular carcinoma of the skin." Its neuroendocrine nature was subsequently clarified, and the discovery in 2008 of the Merkel cell polyomavirus (MCPyV) as a causal agent in approximately 80% of cases in Western populations marked a fundamental advance in understanding its biology.
Etiology: Merkel Cell Polyomavirus (MCPyV) vs UV-Induced MCC
MCC arises through two biologically distinct pathways, distinguishable by the presence or absence of Merkel cell polyomavirus (MCPyV) integration in the tumor genome.
MCPyV-Positive MCC (approximately 80% of cases in the USA and Europe)
MCPyV was discovered in 2008 by Feng et al. and identified as a clonally integrated polyomavirus in the majority of MCC tumors. The mechanism involves monoclonal integration of truncated viral DNA into the host genome — a finding that serves as a molecular fingerprint of viral causation, since all tumor cells from a given patient carry the same integration site. The viral large T-antigen (LT-Ag) and small T-antigen (sT-Ag) function as oncoproteins, disrupting the Rb tumor suppressor pathway and activating translation and cell-cycle progression.
MCPyV is extraordinarily common in the general population — seroprevalence studies suggest that the majority of adults have been exposed by early adulthood, yet MCC develops only rarely. Additional cofactors — particularly immunosuppression and aging — are required for tumor development. In MCPyV-positive tumors, the mutational burden is relatively low, and viral T-antigen epitopes may serve as neoantigens that engage the immune system, which partly explains why these tumors respond well to immune checkpoint inhibitor therapy.
MCPyV-Negative MCC (approximately 20% of cases)
In MCPyV-negative MCC, the tumor arises through a UV-radiation-driven mutational mechanism more closely related to cutaneous squamous cell carcinoma. These tumors carry a high mutational burden, harbor UV-signature C→T transitions, and commonly show TP53 mutations and RB1 deletions. MCPyV-negative MCC is more common in patients of Asian or Pacific Islander ancestry and tends to occur in older, more heavily UV-exposed individuals. The biology overlaps more with carcinogen-driven epithelial cancers than with virus-driven neuroendocrine tumors.
The two subtypes are not always clinically distinguishable at presentation, but the distinction has biological significance: MCPyV serology can be used as a surveillance biomarker only in MCPyV-positive patients, and emerging data suggest differential responses to immune checkpoint inhibitors between the two groups.
Immunosuppression and Risk Factors
The single most important risk factor for MCC beyond age and UV exposure is immunosuppression. The immune system normally surveils and suppresses early MCC development, and any condition that compromises this surveillance dramatically amplifies risk:
- Chronic lymphocytic leukemia (CLL): CLL patients have a 20-fold elevated risk of MCC, the highest relative risk of any immunosuppressive condition. CLL impairs B-cell and T-cell immunity and is thought to permit MCPyV-driven tumor escape. The combination of CLL and MCC represents a particularly aggressive clinical scenario with poor outcomes.
- Solid organ transplantation: Recipients face a 10- to 30-fold increased risk. Maintenance immunosuppression (calcineurin inhibitors, mycophenolate) suppresses the T-cell immunity needed to control MCPyV-positive tumors. MCC in transplant recipients often presents at more advanced stages.
- HIV/AIDS: Approximately 13-fold elevated risk, particularly in patients with uncontrolled viremia and low CD4 counts. Antiretroviral therapy optimization can reduce risk.
- Primary immunodeficiency syndromes: Rare inherited conditions that impair T-cell function (e.g., DOCK8 deficiency) have been reported in association with MCC.
Beyond immunosuppression, established risk factors include:
- UV radiation: Chronic sun exposure, particularly cumulative lifetime UV dose, is the dominant environmental driver. Geographic distribution of MCC follows UV exposure patterns. Tanning bed use has been associated with increased risk. Ozone depletion is believed to contribute to the rising incidence.
- Fair skin phenotype: Fair skin, blue or green eyes, and inability to tan are consistent risk features. MCC is rare in individuals of African descent, likely reflecting the protective effect of melanin.
- Age: The median age at diagnosis is approximately 76 years. MCC under age 50 is rare (less than 5% of cases) and may be associated with a higher prevalence of immunosuppression in younger patients who develop the disease.
- Male sex: Men account for approximately 60% of MCC cases for unclear reasons.
Clinical Presentation
MCC typically presents as a rapidly enlarging, flesh-colored to red-violet firm dermal nodule. The appearance is deceptively benign at initial presentation, often mistaken for a lipoma, inflamed epidermal inclusion cyst, or enlarged lymph node. The HEATH mnemonic captures the characteristic features that should raise suspicion:
- H — Hue: red, violet, or flesh-colored nodule
- E — Expanding rapidly: weeks to months of growth
- A — Asymptomatic: usually painless despite aggressive biology
- T — Tender to palpation in some cases
- H — History of immunosuppression or intense sun exposure
The anatomic distribution reflects UV exposure: approximately 50% of tumors arise on the head and neck (particularly the periorbital region and cheek), 40% on the extremities, and 10% on the trunk. A small fraction arise at sun-protected sites, and rare cases lack a primary cutaneous lesion (presenting with lymph node or visceral MCC alone, known as "unknown primary MCC").
Lymphatic spread is early and clinically silent. Even at presentation without palpable lymphadenopathy — so-called clinically node-negative disease — sentinel lymph node biopsy (SLNB) reveals occult lymph node metastases in approximately 30% of patients. This high rate of occult nodal disease mandates SLNB in virtually all surgically fit patients.
Staging follows the American Joint Committee on Cancer (AJCC) 8th edition system:
- Stage I: Primary tumor ≤2 cm, no nodal involvement — 5-year survival approximately 75%
- Stage II: Primary tumor >2 cm or with local invasion, no nodal involvement — 5-year survival approximately 65%
- Stage III: Regional lymph node involvement (either clinical or microscopic) — 5-year survival approximately 45%
- Stage IV: Distant metastases (most common: liver, lung, bone, brain) — 5-year survival approximately 20%
Diagnosis: Pathology, Immunohistochemistry, and Staging
The diagnosis of MCC requires skin biopsy with histopathologic evaluation and immunohistochemistry (IHC). Clinical suspicion alone is insufficient — MCC mimics many other conditions, and the histology is specific.
Histopathology
On hematoxylin and eosin staining, MCC appears as sheets or trabeculae of small round blue cells within the dermis, often extending into the subcutaneous fat. The cells have scant cytoplasm, finely dispersed ("salt-and-pepper") chromatin, inconspicuous nucleoli, and frequent mitotic figures. Geographic necrosis and lymphovascular invasion are common. The morphologic differential includes metastatic small-cell lung carcinoma, cutaneous lymphoma, Ewing sarcoma, and other small round blue cell tumors — IHC is required for distinction.
Immunohistochemistry
The IHC panel for MCC is distinctive:
- CK20 (cytokeratin 20): Positive in approximately 90–95% of MCC, characteristically in a perinuclear dot-staining pattern — a paranuclear cytoplasmic globule that is nearly pathognomonic of MCC among small round blue cell tumors.
- CAM5.2: Low-molecular-weight cytokeratin, positive in MCC.
- Synaptophysin and chromogranin A: Neuroendocrine markers, positive in most MCC — confirm the neuroendocrine lineage.
- CD56 (NCAM): Also frequently positive.
- CK7: Negative in MCC — the most important distinguishing feature from metastatic small-cell lung carcinoma (SCLC), which is CK7-positive and CK20-negative. The CK7−/CK20+ pattern is the IHC fingerprint of MCC.
- TTF-1: Negative in MCC; positive in SCLC — provides additional distinction from metastatic pulmonary disease.
MCPyV status can be assessed by IHC using the CM2B4 antibody (directed against the large T-antigen) or by quantitative PCR of tumor DNA.
Workup and Staging
Complete staging workup includes:
- SLNB: Mandatory in all surgically fit patients with clinically node-negative disease. PET/CT should be obtained prior to SLNB if available, since PET may identify nodal or distant disease that renders SLNB moot.
- CT chest/abdomen/pelvis: For systemic staging.
- PET/CT: Superior sensitivity for nodal and distant disease; upstages 10–15% of patients beyond conventional CT alone; preferred if available.
- Brain MRI: For patients with neurological symptoms or stage IV disease.
- MCPyV serology (anti-T-antigen antibody titer): Obtained at baseline for all patients; useful as a surveillance biomarker in MCPyV-positive disease.
Treatment: Surgery, Radiation, and Sentinel Lymph Node Biopsy
For localized and regionally confined MCC, treatment combines surgical excision, sentinel lymph node evaluation, and adjuvant radiation therapy. MCC is highly radiosensitive — a property that distinguishes it from many other skin cancers and makes radiation central to both adjuvant and definitive treatment strategies.
Surgery
Wide local excision with 1–2 cm margins is the standard surgical approach. Unlike melanoma, Mohs micrographic surgery does not confer a clear advantage in local control and is not routinely used. The surgical goal is microscopically clear margins; however, local recurrence rates with surgery alone remain high (approximately 40%), which is why adjuvant radiation is nearly universal in patients fit for it.
Sentinel Lymph Node Biopsy
SLNB is performed at the time of or shortly after primary excision. The 30% rate of occult nodal disease in clinically N0 patients makes SLNB the most important staging procedure in MCC. A positive SLNB upstages the patient to Stage III and mandates treatment of the regional nodal basin — either complete lymph node dissection (CLND) or regional radiation therapy (increasingly preferred over CLND due to equivalent regional control and lower morbidity).
Adjuvant Radiation Therapy
Adjuvant radiation to the primary site and regional nodal basin dramatically reduces locoregional recurrence — from approximately 40% to under 10% in most series. Intensity-modulated radiation therapy (IMRT) is preferred for its ability to deliver precise doses while minimizing exposure to surrounding structures. Typical doses are 50–56 Gy to the primary site and 45–50 Gy to regional nodes.
For patients who are medically inoperable, definitive radiation therapy alone achieves excellent local control and represents a legitimate curative-intent strategy for localized MCC.
Adjuvant Systemic Therapy
Platinum-based chemotherapy (carboplatin or cisplatin + etoposide) was historically used in the adjuvant setting but has never demonstrated improved survival in randomized trials and is no longer recommended routinely. Adjuvant immune checkpoint inhibition is under active investigation, with the JAVELIN Merkel 200 extension data showing durable disease-free survival with avelumab in high-risk resected MCC; this remains an evolving area and is not yet universally incorporated into guidelines.
Immunotherapy: Avelumab, Pembrolizumab, and the Revolution in MCC
The development of immune checkpoint inhibitors targeting the PD-1/PD-L1 axis transformed MCC from a disease with dismal outcomes in the metastatic setting to one in which durable complete remissions are achievable in a meaningful fraction of patients. MCC was, in many ways, an ideal tumor for immunotherapy: it carries viral neoantigens (in MCPyV-positive cases), often expresses PD-L1, and is associated with states of immune deficiency that checkpoint inhibition can potentially reverse.
Pembrolizumab (KEYTRUDA)
Pembrolizumab — a humanized anti-PD-1 monoclonal antibody — received FDA approval in December 2018 for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic MCC based on the KEYNOTE-017 trial. In this single-arm phase 2 trial of 50 patients with advanced MCC who had not received prior systemic therapy, pembrolizumab achieved an overall response rate (ORR) of 56% at 24 months, including 24% complete responses. Responses were durable — the median duration of response had not been reached at 24-month follow-up, and 69% of responses were ongoing at that timepoint. Pembrolizumab is now the preferred first-line agent for advanced MCC at most centers.
Avelumab (BAVENCIO)
Avelumab — an anti-PD-L1 antibody — received FDA approval in March 2017, becoming the first immunotherapy approved for MCC and the first anti-PD-L1 agent approved for any cancer. Approval was based on the JAVELIN Merkel 200 trial, a phase 2 study of 88 patients with chemotherapy-refractory metastatic MCC. Avelumab achieved a confirmed ORR of 33%, with 11% complete responses, and a 12-month progression-free survival (PFS) of 30%. Duration of response was durable in responding patients.
Nivolumab
Nivolumab (anti-PD-1) demonstrated an ORR of 68% in the first-line setting in the CheckMate 358 trial (Cohort B), with similarly durable responses. Though not FDA-approved for MCC, it provides an additional anti-PD-1 option and is used off-label at some centers.
Comparison with Prior Chemotherapy
Prior to immunotherapy, first-line systemic treatment for advanced MCC consisted of platinum-based doublet chemotherapy (carboplatin or cisplatin + etoposide). Response rates of 50–60% were achieved, but responses were almost uniformly transient, with a median duration of approximately 3 months and no demonstrated improvement in overall survival. Chemotherapy is now reserved for immunotherapy-refractory patients or situations where rapid debulking is needed while awaiting immunotherapy response.
Biology of Response
MCPyV-positive tumors may respond better to checkpoint inhibition because viral T-antigen peptides serve as tumor-specific neoantigens, generating a pre-existing immune response that checkpoint blockade can amplify. MCPyV-negative tumors, with their higher mutational burden, also respond but outcomes may be somewhat more heterogeneous. PD-L1 expression on tumor cells is common in MCC and correlates with response in some analyses, but is not routinely used to select patients for therapy.
Prognosis, Surveillance, and MCPyV Serology as Biomarker
MCC has a high recurrence rate — approximately 40–50% of patients experience disease recurrence, most commonly within the first two years after diagnosis. Locoregional recurrence (local skin, in-transit lesions, regional nodes) is the most common pattern in early-stage disease; distant recurrence affects the liver, lung, bone, and brain in advanced cases.
MCPyV Antibody Titer as a Surveillance Biomarker
One of the most valuable and novel features of MCC — unique among skin cancers — is the use of MCPyV oncoprotein antibody titers as a real-time biomarker of tumor burden in MCPyV-positive patients. Antibodies against the MCPyV small T-antigen and large T-antigen (measurable using the AMERK assay) are detectable at baseline in the majority of MCPyV-positive MCC patients. Key clinical principles:
- A high titer at diagnosis indicates active MCPyV-positive tumor and establishes a baseline for monitoring.
- A falling titer after treatment correlates with effective tumor clearance.
- A rising titer during surveillance is a sensitive early signal of recurrence that can precede radiographic or clinical detection by weeks to months, enabling earlier intervention.
- Patients who are MCPyV-seronegative at diagnosis either have MCPyV-negative tumors or are immunologically unable to mount an antibody response; serology cannot be used for surveillance in these patients.
Surveillance Schedule
Standard post-treatment surveillance includes:
- History and physical examination every 3–6 months for the first 3 years, then annually thereafter
- Cross-sectional imaging (CT chest/abdomen/pelvis or PET/CT) every 6–12 months for 3 years
- MCPyV antibody titers at each visit in seropositive patients
- Skin examination at each visit, with particular attention to the primary site, in-transit lymphatics, and regional nodes
Distant recurrence beyond 5 years is uncommon but documented; lifelong awareness of recurrence risk is warranted for survivors.
Special Populations: Immunocompromised Patients
Immunocompromised patients represent the highest-risk subgroup in MCC and pose distinct management challenges. They account for a disproportionate fraction of MCC cases and face uniformly worse outcomes compared to immunocompetent patients:
- More advanced stage at presentation
- Higher rates of locoregional and distant recurrence
- Shorter disease-specific and overall survival
- Reduced immune response to checkpoint inhibitor therapy in some subgroups
Solid Organ Transplant Recipients
Checkpoint inhibitor use in transplant recipients carries significant risk of allograft rejection, mediated by T-cell activation against both tumor and allograft. Case reports and series document rates of rejection of 40–50% in solid organ transplant recipients receiving anti-PD-1/PD-L1 therapy. Management requires careful multidisciplinary discussion between oncology and transplant medicine. Reduction of immunosuppression — if clinically feasible without threatening the allograft — may improve immune surveillance. In some patients, the risk-benefit calculation favors immunotherapy despite transplant risk; in others, radiation and surgery without systemic immunotherapy may be the preferred approach.
Patients with CLL
CLL + MCC is a particularly aggressive combination. CLL-directed therapies, particularly BTK inhibitors (ibrutinib, acalabrutinib), may inadvertently modulate immune function in ways that affect MCC biology. Ibrutinib has complex immunologic effects — it suppresses some pathologic B-cell activity while potentially impairing T-cell function. Whether CLL-directed therapy improves or worsens MCC outcomes in individual patients requires careful case-by-case assessment. Checkpoint inhibitors may be used in CLL + MCC but clinical trial data specific to this population are limited.
HIV-Positive Patients
In HIV-positive patients, optimization of antiretroviral therapy to achieve viral suppression and immune reconstitution is a critical adjunct to MCC-directed therapy. Immune reconstitution can restore the T-cell surveillance that normally suppresses MCPyV-driven tumor growth, and some patients have experienced tumor regression with ART alone. Checkpoint inhibitors appear to be safe in HIV-positive patients with well-controlled viral loads, though prospective data in MCC specifically are limited.
Key Research Papers
- Feng H et al. "Clonal integration of a polyomavirus in human Merkel cell carcinoma." Science. 2008;319(5866):1096–100. PMID 18202256
- Nghiem PT et al. "PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma." N Engl J Med. 2016;374(26):2542–52. PMID 27093365
- Kaufman HL et al. "Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma." Lancet Oncol. 2016;17(10):1374–85. PMID 27592805
- Becker JC et al. "Merkel cell carcinoma." Nat Rev Dis Primers. 2017;3:17077. — Search PubMed
- Nghiem P et al. "Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy." J Clin Oncol. 2019;37(9):693–702. — Search PubMed
- D'Angelo SP et al. "Nivolumab in Merkel Cell Carcinoma, Checkpoint Inhibition in Patients with Merkel Cell Carcinoma." J Clin Oncol. 2018;36(28):2834–2838. — Search PubMed
- Lemos B et al. "Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma." J Am Acad Dermatol. 2010;63(5):751–61. — Search PubMed
- Strom T et al. "Regional Radiation Therapy Reduces Disease Recurrence and Improves Disease-Specific Survival in Patients with Merkel Cell Carcinoma of the Lower Extremity." Ann Surg Oncol. 2016;23(9):2854–61. — Search PubMed
- Paulson KG et al. "Use of Merkel cell polyomavirus antibodies in clinical monitoring of Merkel cell carcinoma patients." J Clin Oncol. 2010;28(9):1582–8. — Search PubMed
- Albores-Saavedra J et al. "Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases." Ann Diagn Pathol. 2010;14(5):301–13. — Search PubMed
- Pectasides D et al. "Merkel cell carcinoma: a systematic review and meta-analysis." Anticancer Res. 2006;26(6C):4841–9. — Search PubMed
- Tilling T et al. "Merkel Cell Carcinoma: Advances in Understanding of Molecular Biology, Clinical Diagnosis and Targeted Therapy." Front Oncol. 2020;10:185. — Search PubMed
Connections
- Oncology
- Skin Cancer
- Neuroendocrine Tumors
- Primary CNS Lymphoma
- Uveal Melanoma
- Chronic Lymphocytic Leukemia
- Lymphoma