Hypereosinophilic Syndrome
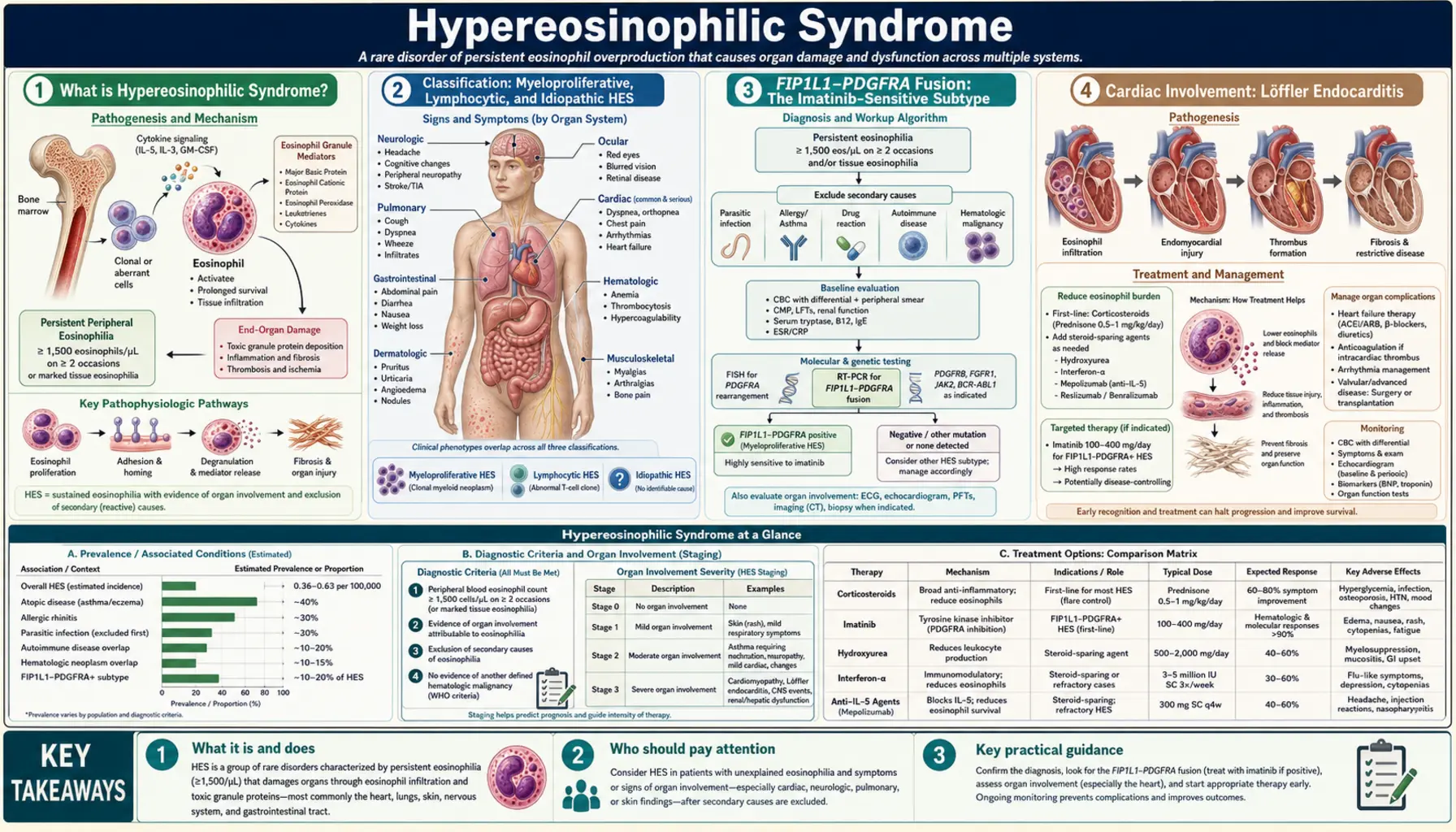
Table of Contents
- What is Hypereosinophilic Syndrome?
- Classification: Myeloproliferative, Lymphocytic, and Idiopathic HES
- FIP1L1-PDGFRA Fusion: The Imatinib-Sensitive Subtype
- Cardiac Involvement: Löffler Endocarditis
- Clinical Presentation and Organ Damage
- Diagnosis and Workup
- Treatment: Imatinib, Mepolizumab, and Corticosteroids
- Long-Term Outcomes and Monitoring
- Research Papers
- Connections
- Featured Videos
What is Hypereosinophilic Syndrome?
Hypereosinophilic Syndrome (HES) is a rare group of disorders defined by persistent, marked eosinophilia in the peripheral blood — an absolute eosinophil count (AEC) of 1,500 per microliter or greater, sustained for at least one month — combined with evidence of eosinophil-mediated damage to one or more organs. The organs most commonly affected are the heart, skin, lungs, nervous system, and gastrointestinal tract. HES is distinguished from reactive eosinophilia (caused by parasites, allergies, or medications) by the absence of an identifiable secondary cause and the direct demonstration of eosinophil-driven tissue injury.
Eosinophils are normally a minor component of white blood cells — typically less than 500 per microliter and making up roughly 1–3% of circulating leukocytes. In HES, this count can rise to tens of thousands per microliter. At such levels, eosinophils migrate into tissues and release toxic granule contents — including major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), and eosinophil peroxidase (EPO) — directly damaging cell membranes, nerves, and cardiac muscle. The resulting organ damage, particularly to the heart, accounts for the historically high morbidity and mortality of HES before modern targeted therapies were available.
HES is classified into three main subtypes based on whether the eosinophilia arises from a clonal myeloid proliferation, an aberrant T-cell clone driving cytokine overproduction, or neither identifiable mechanism. This distinction is critical because subtype determines treatment — myeloproliferative HES driven by the FIP1L1-PDGFRA fusion gene responds dramatically to imatinib, while lymphocytic HES responds to corticosteroids and the anti-IL-5 biologic mepolizumab. Accurate subtyping can be lifesaving.
Classification: Myeloproliferative, Lymphocytic, and Idiopathic HES
The 2022 World Health Organization classification and international consensus guidelines recognize three principal variants of HES, each with distinct biology, clinical features, and treatment approaches.
Myeloproliferative HES (M-HES): Clonal Eosinophilia
M-HES is caused by a clonal abnormality in a hematopoietic stem cell that drives autonomous overproduction of eosinophils. The most important — and most common — molecular driver is the FIP1L1-PDGFRA fusion gene, arising from an interstitial deletion on chromosome 4q12. This fusion creates a constitutively active tyrosine kinase that drives uncontrolled proliferation of eosinophil precursors. Additional clonal markers include PDGFRB rearrangements (e.g., ETV6-PDGFRB), FGFR1 rearrangements, JAK2 mutations, and BCR-ABL-like fusions in rare cases. M-HES predominantly affects men (male-to-female ratio approximately 9:1 for FIP1L1-PDGFRA), with a median age at diagnosis in the 40s. Serum tryptase is characteristically elevated, reflecting a concurrent mast cell component (mast cells and eosinophils share a common progenitor affected by the fusion). Endomyocardial fibrosis and thrombosis are particularly frequent in FIP1L1-PDGFRA-positive M-HES.
Lymphocytic HES (L-HES): Cytokine-Driven Eosinophilia
L-HES is driven not by a myeloid clone but by an aberrant T-cell clone that overproduces interleukin-5 (IL-5), the key cytokine driving eosinophil production, survival, and activation. The abnormal T-cell population typically displays an anomalous immunophenotype on flow cytometry — most often CD3-negative/CD4-positive (lacking the CD3 T-cell receptor complex normally required for surface CD4 expression), or other aberrant combinations — detectable by peripheral blood flow cytometry. T-cell receptor gene rearrangement studies confirm clonality. Skin involvement (urticaria, angioedema, erythroderma, pruritic papules) is the predominant organ manifestation in L-HES and often precedes the diagnosis by years. Serum IgE is markedly elevated in many L-HES patients. The critical long-term concern in L-HES is transformation to overt T-cell lymphoma — the clonal T-cell population, if left unchecked, can evolve into full malignancy, requiring long-term monitoring with periodic bone marrow evaluation and T-cell clonality reassessment.
Idiopathic HES
Idiopathic HES is a diagnosis of exclusion — persistent eosinophilia with organ damage, but with no identifiable clonal (myeloid or lymphoid) driver after comprehensive evaluation. As molecular diagnostic techniques advance, some cases previously labeled idiopathic are being reclassified as M-HES or L-HES. Treatment of idiopathic HES is guided by the degree and nature of organ involvement, typically using corticosteroids as first-line therapy with the anti-IL-5 biologic mepolizumab as a steroid-sparing option.
FIP1L1-PDGFRA Fusion: The Imatinib-Sensitive Subtype
The discovery of the FIP1L1-PDGFRA fusion gene in 2003 transformed the treatment of myeloproliferative HES and stands as one of the most important advances in hematology in the past two decades. This fusion arises from an interstitial deletion of approximately 800 kilobases on chromosome 4q12, joining the FIP1L1 gene (FIP1-like 1, a cleavage and polyadenylation factor) to the PDGFRA gene (platelet-derived growth factor receptor alpha). The resulting chimeric protein retains the tyrosine kinase domain of PDGFRA — now constitutively active because the fusion removes an autoinhibitory domain present in the normal receptor — and drives relentless eosinophil overproduction.
Imatinib: Highly Effective and Rapid
The FIP1L1-PDGFRA tyrosine kinase is exquisitely sensitive to imatinib (Gleevec), the BCR-ABL inhibitor originally developed for chronic myeloid leukemia. Doses as low as 100 mg per day — far below the standard 400 mg dose used for CML — produce complete hematologic remissions in more than 95% of FIP1L1-PDGFRA-positive patients, typically within four to eight weeks. Molecular remissions (undetectable fusion transcript by RT-PCR) are achieved in the majority of patients on maintenance therapy. The response is durable as long as treatment continues; discontinuation is cautiously attempted in patients with sustained deep molecular remission, though relapse is common.
Diagnosis of FIP1L1-PDGFRA
The fusion is not detected by standard chromosomal karyotyping because the deleted segment is too small to visualize on conventional cytogenetics. Diagnosis requires fluorescence in situ hybridization (FISH) using probes flanking the CHIC2 locus on chromosome 4q12 (deleted in the fusion) or reverse-transcription PCR (RT-PCR) to detect the fusion transcript. All patients with suspected M-HES or unexplained persistent eosinophilia should undergo FISH/RT-PCR testing specifically for FIP1L1-PDGFRA before treatment decisions are made, as this finding mandates imatinib therapy and spares patients from unnecessary immunosuppression with its attendant toxicities.
Cardiac Involvement: Löffler Endocarditis
Cardiac involvement is the most feared complication of HES and the primary driver of morbidity and mortality before effective therapies were available. The cardiac disease in HES — historically called Löffler endocarditis (named after Swiss physician Wilhelm Löffler who described the syndrome in the 1930s and 1940s) — is caused by the direct toxicity of eosinophil granule proteins on cardiac tissue and proceeds through three well-defined stages.
Stage 1: Acute Necrotic Phase
In the acute phase, eosinophils infiltrate the myocardium and release their granule contents — particularly major basic protein (MBP) and eosinophil cationic protein (ECP) — directly into cardiac tissue. These highly positively-charged proteins intercalate into cell membranes, disrupt ion channel function, and trigger apoptosis of myocardial cells. The result is focal myocardial necrosis, with T-cell-mediated inflammatory infiltration contributing to tissue damage. Clinically, this phase may manifest as acute myocarditis with chest pain, dyspnea, and elevated troponin — or may be entirely subclinical, detectable only by cardiac biomarkers or cardiac MRI.
Stage 2: Thrombotic Phase
As the necrotic myocardium heals, mural thrombi form on the damaged endomyocardial surface, particularly at the ventricular apices. These thrombi are a major source of systemic thromboembolism — embolic strokes, peripheral arterial occlusion, and mesenteric ischemia are all recognized complications of HES cardiac involvement. Anticoagulation during this phase reduces embolic risk. Echocardiography typically shows increased echogenicity at the ventricular apices, filling defects consistent with thrombus, and variable degrees of valvular dysfunction.
Stage 3: Fibrotic Phase
The final and most advanced stage is characterized by fibrosis replacing the damaged and thrombotic endomyocardium. Progressive endomyocardial fibrosis causes restrictive cardiomyopathy — the heart walls become stiff and non-compliant, impairing diastolic filling and ultimately leading to biventricular heart failure. Fibrosis may also tether or distort the mitral and tricuspid valve apparatus, causing regurgitation from papillary muscle involvement. By this stage, the damage is largely irreversible. Surgical treatment (endocardial resection and valve repair) may be required in advanced fibrotic Löffler cardiomyopathy. Echocardiography in the fibrotic phase shows ventricular apex obliteration — the apices become filled with dense fibrotic tissue — a nearly pathognomonic appearance.
Cardiac Screening
Because early cardiac involvement can be silent and because intervention before fibrosis is far more effective, all patients with HES should undergo baseline echocardiography and cardiac troponin testing at diagnosis, with repeat echocardiography every 6–12 months during active disease. Cardiac MRI with gadolinium late enhancement is more sensitive than echo for detecting early myocarditis and fibrosis and should be performed when cardiac involvement is suspected but echocardiography is inconclusive.
Clinical Presentation and Organ Damage
The clinical presentation of HES is highly variable and reflects the organ systems infiltrated by eosinophils. The diagnosis is frequently delayed because early manifestations (urticaria, cough, fatigue) are nonspecific and attributed to common conditions. A high index of suspicion is warranted whenever persistent eosinophilia is discovered.
Skin Involvement
Cutaneous manifestations are the most common non-cardiac feature of HES and are the dominant presentation in L-HES. Findings include urticaria (hives), angioedema, erythematous pruritic papules and nodules, erythroderma (generalized skin redness), and mucosal ulcers. These skin findings can resemble allergic disease or chronic urticaria and may precede the diagnosis of HES by years. Unlike allergic urticaria, HES-related skin involvement tends to be chronic, poorly responsive to antihistamines, and recurrent despite avoiding identifiable triggers.
Pulmonary Involvement
Eosinophilic infiltration of the lungs causes cough, dyspnea, wheeze, and exertional limitation. Imaging may show eosinophilic pneumonia-pattern infiltrates (migratory peripheral consolidations) or interstitial changes. Pulmonary function testing often demonstrates a restrictive or mixed pattern. Pleural effusions may occur. Pulmonary involvement is usually responsive to corticosteroids, but recurs with dose reduction in HES.
Neurological Involvement
Neurological manifestations include peripheral neuropathy (sensorimotor, from direct eosinophil infiltration of endoneurium and perineurium), embolic strokes (from cardiac thrombi — see cardiac section), encephalopathy, and cognitive impairment. In severe cases, eosinophilic meningitis may occur. Peripheral neuropathy in HES can be painful and debilitating, often affecting the distal lower extremities asymmetrically.
Gastrointestinal Involvement
Eosinophilic involvement of the GI tract causes abdominal pain, diarrhea (including bloody diarrhea), nausea, vomiting, and malabsorption. Any level of the GI tract from esophagus to colon may be affected. Ascites from eosinophilic serositis may occur. Hepatomegaly and liver function abnormalities are found in some patients.
Diagnosis and Workup
The diagnostic approach to a patient with eosinophilia ≥1,500/µL requires two parallel tracks: (1) ruling out secondary (reactive) causes of eosinophilia, and (2) classifying the subtype of HES once reactive causes are excluded.
Ruling Out Secondary Eosinophilia
Secondary eosinophilia must be rigorously excluded before diagnosing HES, because the treatment approaches are entirely different. The most common secondary causes include:
- Parasitic infections: Stool ova and parasite (O&P) examination × 3 specimens; serologic testing for Strongyloides stercoralis (most important — risk of fatal hyperinfection with corticosteroids if missed), Toxocara canis/cati, Trichinella spiralis, and filarial species in patients from endemic regions.
- Drug reaction: Comprehensive medication history including supplements and over-the-counter agents; drug-induced eosinophilia (DRESS syndrome) can reach HES-range counts.
- Allergic disease: Serum IgE; skin-prick or RAST testing if relevant.
- Vasculitis: ANCA (anti-neutrophil cytoplasmic antibody) panel — eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg-Strauss) is an important mimic with both eosinophilia and vasculitic organ damage.
- Solid tumors and lymphomas: CT scan of chest/abdomen/pelvis to identify occult malignancy.
- Adrenal insufficiency: Morning cortisol (adrenal insufficiency causes eosinophilia and can mimic HES symptoms).
Subtyping HES
Once reactive causes are excluded, classification into M-HES, L-HES, or idiopathic requires:
- FISH and/or RT-PCR for FIP1L1-PDGFRA (mandatory in all cases; positive result = M-HES and imatinib-sensitive)
- Conventional cytogenetics (to detect PDGFRB rearrangements, FGFR1 fusions, or other clonal abnormalities by standard karyotype and FISH panels)
- Bone marrow biopsy with aspirate (cellularity; eosinophil precursors; blast percentage; cytogenetics; mast cell component)
- Peripheral blood flow cytometry (T-cell immunophenotyping — detect aberrant CD3-/CD4+ or other anomalous T-cell populations)
- T-cell receptor gene rearrangement studies (confirm T-cell clonality in suspected L-HES)
- Serum tryptase (elevated in FIP1L1-PDGFRA-positive M-HES; supports mast cell involvement)
- Serum IgE (markedly elevated in L-HES)
- Echocardiography + troponin (baseline cardiac assessment; repeat every 6–12 months)
Treatment: Imatinib, Mepolizumab, and Corticosteroids
Treatment is subtype-directed and severity-dependent. Patients with imminent or active life-threatening organ compromise (acute myocarditis, rapidly worsening respiratory failure, stroke) may require immediate eosinophil cytoreduction with high-dose corticosteroids or hydroxyurea while diagnostic evaluation is completed.
FIP1L1-PDGFRA-Positive M-HES: Imatinib
Imatinib (Gleevec) is the standard of care for FIP1L1-PDGFRA-positive HES. Starting at 100 mg per day (not the 400 mg used for CML), imatinib produces complete hematologic remissions (eosinophil count normalized) in more than 95% of patients, typically within 4–8 weeks. Molecular remissions follow in the majority of patients on sustained therapy. Imatinib is generally well tolerated at the low doses used for HES; common side effects include edema, fatigue, nausea, and muscle cramps. Patients should be monitored by FIP1L1-PDGFRA RT-PCR every 3–6 months; persistent molecular remission for ≥2 years may allow cautious dose tapering with very close monitoring. Resistance to imatinib in FIP1L1-PDGFRA HES is rare but has been reported; in such cases, second-generation TKIs (sorafenib) have shown activity.
Corticosteroids: Broad First-Line for Non-PDGFRA HES
Prednisone (or methylprednisolone for severe disease) is the cornerstone initial therapy for L-HES and idiopathic HES when FIP1L1-PDGFRA has been excluded. The typical starting dose is 1 mg/kg/day (up to 60 mg/day), producing rapid eosinophil reduction in most patients within 24–48 hours. Corticosteroids work by suppressing IL-5 signaling and directly driving eosinophil apoptosis. However, most patients require prolonged corticosteroid therapy, and the cumulative toxicity (weight gain, osteoporosis, diabetes, cataracts, adrenal suppression, infections) makes steroid-sparing strategies essential.
Mepolizumab: FDA-Approved Anti-IL-5 Therapy
Mepolizumab (Nucala), a humanized monoclonal antibody targeting interleukin-5, received FDA approval for HES in 2020 — the first biologic specifically approved for this indication. By neutralizing IL-5, mepolizumab dramatically reduces eosinophil production, survival, and activation. The landmark COMET trial demonstrated that mepolizumab 300 mg subcutaneously every 4 weeks significantly reduced HES flares and allowed corticosteroid dose reduction or discontinuation in the majority of patients. Mepolizumab is now considered a cornerstone steroid-sparing therapy for FIP1L1-PDGFRA-negative HES and is particularly valuable in L-HES. Dupilumab (anti-IL-4/IL-13) is an emerging option for L-HES with high IgE.
Additional Agents
Hydroxyurea provides cytoreduction in M-HES or severe idiopathic HES when faster eosinophil reduction is needed than corticosteroids alone can provide. Interferon-alfa has activity in L-HES and idiopathic HES, particularly as a corticosteroid-sparing agent; it is less convenient (injections, significant side-effect profile) but remains a valuable second-line option. Anticoagulation (typically with heparin bridging to warfarin or a DOAC) is indicated for HES patients with documented cardiac thrombus during the thrombotic phase of Löffler endocarditis. Allogeneic stem cell transplantation is reserved for refractory cases, particularly those transforming to acute leukemia or in younger patients with high-risk clonal disease unresponsive to available therapies.
Long-Term Outcomes and Monitoring
The prognosis of HES has improved dramatically with the advent of targeted therapies. FIP1L1-PDGFRA-positive M-HES, once frequently fatal from cardiac disease, now carries an excellent prognosis with sustained imatinib therapy. Patients require lifelong monitoring because eosinophilia can recur with treatment interruption and cardiac damage, once established, is largely irreversible.
L-HES patients require long-term surveillance for T-cell lymphoma transformation — periodic T-cell receptor rearrangement studies and bone marrow biopsy are recommended every 1–2 years. The risk of lymphoma is estimated at 10–25% over a decade of observation in clonal L-HES, though not all series agree. Early lymphoma transformation may be detected as worsening eosinophilia, lymph node enlargement, B-symptoms, or a change in the aberrant T-cell clone's immunophenotype.
All patients with HES should have annual echocardiography, complete blood counts every 3–6 months during stable disease, and careful clinical reassessment for new or worsening organ involvement. Quality of life in well-controlled HES is often very good, but treatment-related toxicities (long-term corticosteroids, imatinib side effects) require ongoing management.
Research Papers
- Gotlib J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97(1):129–148 — Search PubMed
- Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201–1214. PMID: 12660384
- Klion AD, Bochner BS, Gleich GJ, et al. Approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J Allergy Clin Immunol. 2006;117(6):1292–1302. PMID: 16750989
- Rothenberg ME, Klion AD, Roufosse FE, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med. 2008;358(12):1215–1228. PMID: 18344568
- Klion AD. How I treat hypereosinophilic syndromes. Blood. 2015;126(9):1069–1077 — Search PubMed
- Roufosse F, Weller PF. Practical approach to the patient with hypereosinophilia. J Allergy Clin Immunol. 2010;126(1):39–44 — Search PubMed
- Ogbogu PU, Bochner BS, Butterfield JH, et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol. 2009;124(6):1319–1325. PMID: 19910029
- Roufosse FE, Kahn JE, Gleich GJ, et al. Long-term safety of mepolizumab for the treatment of hypereosinophilic syndromes. J Allergy Clin Immunol. 2013;131(2):461–467 — Search PubMed
- Legrand F, Renneville A, Macintyre E, et al. The spectrum of FIP1L1-PDGFRA-associated chronic eosinophilic leukemia: new insights based on a survey of 44 cases. Medicine. 2013;92(1):e1–e9 — Search PubMed
- Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann Intern Med. 1968;68(6):1220–1229 — Search PubMed
- Gleich GJ, Schroeter AL, Marcoux JP, et al. Episodic angioedema associated with eosinophilia. N Engl J Med. 1984;310(25):1621–1626 — Search PubMed
- Klion AD, Noel P, Akin C, et al. Elevated serum tryptase levels identify a subset of patients with a myeloproliferative variant of idiopathic hypereosinophilic syndrome associated with tissue fibrosis, poor prognosis, and imatinib responsiveness. Blood. 2003;101(12):4660–4666. PMID: 12676775
Search PubMed for more: Hypereosinophilic syndrome FIP1L1-PDGFRA imatinib Mepolizumab HES
Connections
- Hematology
- Mastocytosis
- Essential Thrombocythemia
- Chronic Lymphocytic Leukemia
- Primary Myelofibrosis
- Myelodysplastic Syndrome
- Non-Hodgkin Lymphoma
- Complete Blood Count