Rett Syndrome
- Overview and Epidemiology
- The MECP2 Gene and Epigenetic Mechanism
- Why Females Survive: X-Inactivation
- Clinical Stages of Classic Rett Syndrome
- Diagnosis
- Seizures and Breathing Irregularities
- Cardiac Risks and Monitoring
- Management and Supportive Care
- Trofinetide (DAYBUE) — FDA-Approved 2023
- Gene Therapy and Future Directions
- Key Research Papers
- Connections
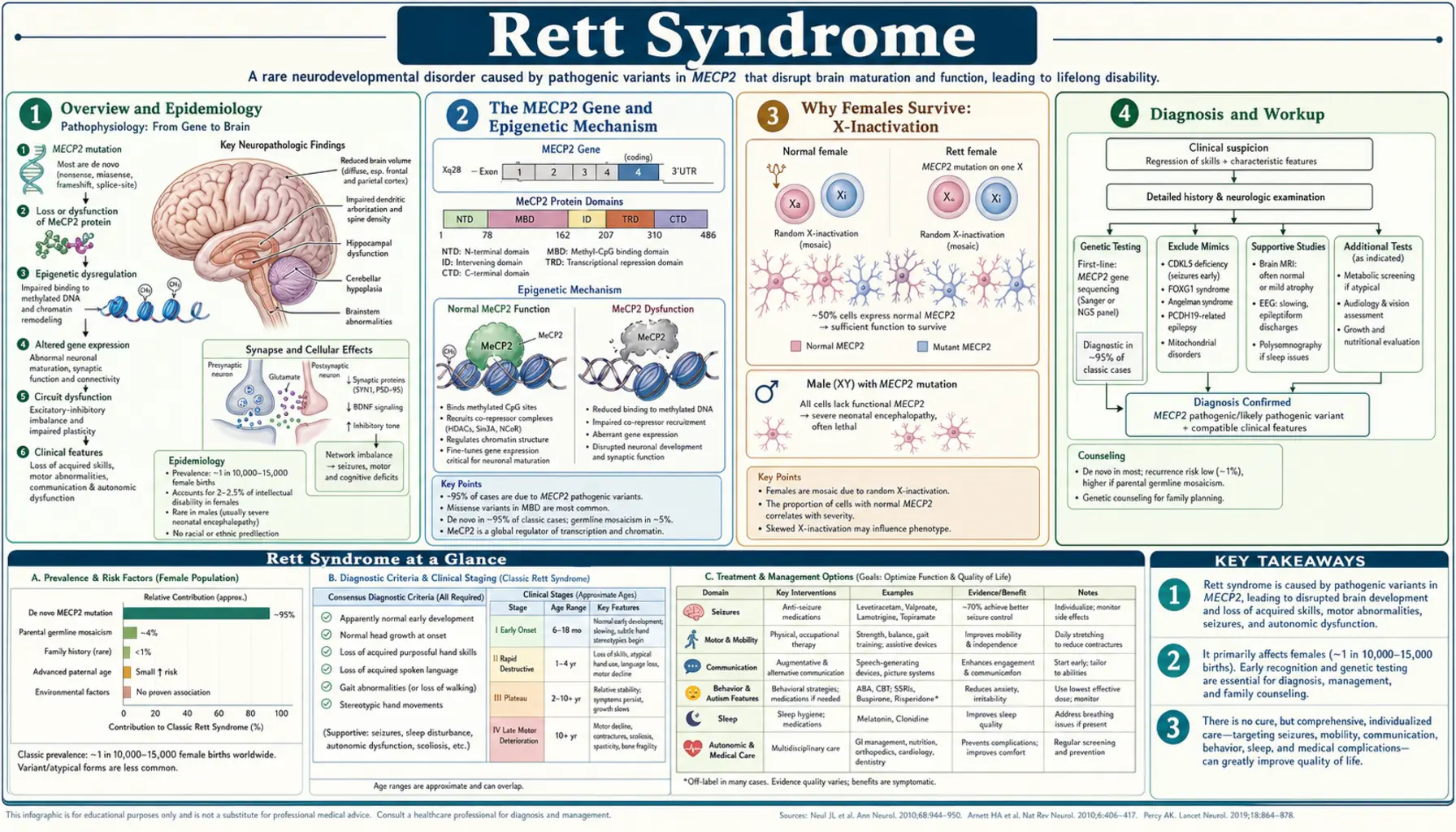
Overview and Epidemiology
Rett syndrome (RTT) is a severe X-linked dominant neurodevelopmental disorder that affects almost exclusively females. It is one of the most common genetic causes of profound intellectual disability in girls worldwide. Unlike many genetic conditions that are apparent from birth, Rett syndrome has a deceptive early course — affected girls appear to develop normally for the first six to eighteen months of life, then enter a period of rapid and devastating regression that strips away the language, purposeful hand use, and social skills they had already gained.
The condition was first described in 1966 by Austrian pediatric neurologist Andreas Rett, who noticed a distinctive pattern of hand-wringing stereotypies and developmental regression in two girls sitting side by side in his waiting room. His original German-language reports were largely overlooked until 1983, when Swedish child neurologist Bengt Hagberg and colleagues published an English-language description of 35 cases in Annals of Neurology, bringing Rett syndrome to international attention.
The genetic cause — mutations in the MECP2 gene on the X chromosome — was identified in 1999 by Ruthie Amir and Huda Zoghbi at Baylor College of Medicine, transforming Rett syndrome from a clinical mystery into one of the best-understood epigenetic disorders in neuroscience.
How Common Is Rett Syndrome?
Rett syndrome affects approximately 1 in 10,000 to 15,000 females. In the United States, this translates to roughly 6,000 to 9,000 affected individuals. Because it is an X-linked dominant condition and males with the typical mutation do not survive to birth or die in early infancy, essentially all patients are female. The condition occurs in all racial and ethnic groups at similar rates, with no geographic clustering, reflecting the fact that most cases arise from new (de novo) mutations rather than inherited ones. More than 99% of cases are sporadic — a daughter with Rett syndrome almost never has a mother with the condition.
The MECP2 Gene and Epigenetic Mechanism
The MECP2 gene sits at chromosome Xq28 — near the tip of the long arm of the X chromosome — and encodes methyl-CpG-binding protein 2 (MeCP2). Understanding what MeCP2 does at the molecular level is essential to understanding why its loss causes such widespread neurological damage.
What Does MeCP2 Do?
MeCP2 is an epigenetic reader and interpreter. DNA methylation — the attachment of a methyl group to cytosine bases, especially at CpG dinucleotides — is one of the primary ways the genome marks genes for silencing. MeCP2 reads these methylation marks: it binds specifically to methylated CpG sites throughout the genome and recruits co-repressor complexes that lock those genes down. Think of DNA methylation as a "do not express" sticky note on a gene, and MeCP2 as the protein that reads that sticky note and calls in the silencing machinery.
In mature neurons, MeCP2 is expressed at exceptionally high levels — higher than in almost any other cell type. Neurons use MeCP2 constantly to suppress genes that should stay silent after the brain has finished its early development. When MeCP2 is lost or dysfunctional, those silencing signals are not enforced: genes that should remain quiet become inappropriately active in mature neurons, producing proteins that disrupt the finely tuned balance of neuronal circuits.
Brain Changes in Rett Syndrome
Post-mortem studies of Rett syndrome brains reveal several consistent findings: reduced brain weight (particularly in the frontal cortex and motor cortex), decreased synapse density, and abnormalities in dendritic branching. Neurons are smaller and more densely packed than normal — a pattern suggesting arrested maturation rather than cell death. The neurotransmitter systems most affected include dopamine, serotonin, and glutamate signaling pathways, which explains the broad clinical spectrum: movement abnormalities, mood dysregulation, seizures, and autonomic dysfunction all flow from disruptions in these interconnected systems.
Types of MECP2 Mutations
Over 200 distinct mutations in MECP2 have been associated with Rett syndrome, but eight recurring mutations account for approximately two-thirds of all cases. These are designated by the amino acid change they cause: R106W, R133C, T158M, R168X, R255X, R270X, R294X, and R306C. The letter-number combinations refer to the position in the protein and the amino acid substitution (e.g., R255X means the arginine at position 255 is replaced by a stop codon, truncating the protein).
Genotype-phenotype correlations — the relationship between which mutation a girl has and how severely she is affected — exist but are imperfect. In general, mutations that truncate the protein early (such as R168X) tend to produce more severe phenotypes, while mutations that preserve the methyl-binding domain (such as R294X or R306C) are sometimes associated with milder disease. However, the same mutation can produce substantially different severity in different girls, largely because of differences in X-inactivation patterns (see below).
Why Females Survive: X-Inactivation
One of the most fundamental questions about Rett syndrome is: why do girls survive when boys with the same mutation do not? The answer lies in a cellular process called X-inactivation (also called lyonization, after geneticist Mary Lyon who described it in 1961).
How X-Inactivation Works
Human females carry two X chromosomes — one from each parent. Early in embryonic development, each cell independently and randomly silences one of its two X chromosomes. From that point forward, every descendant of that cell carries the same silenced X. The result is that a normal adult female is a mosaic: roughly half her cells express genes from the maternal X chromosome, and the other half express genes from the paternal X chromosome. This mosaicism is permanent and is reflected in traits like the patchy coat color of calico cats.
Why Mosaicism Is Life-Saving in RTT
In a girl who carries an MECP2 mutation on one X chromosome, X-inactivation creates a cellular mosaic: approximately 50% of her neurons carry the mutant MECP2 allele on the active X (and therefore produce dysfunctional MeCP2), while the other 50% carry the normal allele on the active X (and produce functional MeCP2). This mixture of normal and abnormal neurons is what makes survival possible — the normal neurons provide enough MeCP2 function to sustain basic life, while the abnormal neurons cause the progressive neurological disorder.
Males, with only a single X chromosome, cannot have this mosaic rescue. A boy who inherits a classic RTT-causing MECP2 mutation has no cells producing normal MeCP2. The result is either fetal lethality (most affected male conceptuses do not survive to birth) or, in rare cases where the mutation is less severe, a devastating neonatal encephalopathy with hypotonia, respiratory failure, and early death. The only exceptions are males who are mosaic for the mutation (it arose post-fertilization, so some cells are unaffected) or males with Klinefelter syndrome (XXY) who, like females, have one X inactivated.
X-Inactivation Skewing
In some girls with Rett syndrome, X-inactivation is not 50:50 — it is "skewed," meaning a disproportionate fraction of cells may silence one X over the other. Favorable skewing (where the mutant X is preferentially silenced in more cells) can result in a milder phenotype; unfavorable skewing (where the normal X is more often silenced) can worsen outcomes. This is why two girls with identical MECP2 mutations can have significantly different clinical courses. Researchers are actively exploring whether X-inactivation skewing can be therapeutically manipulated.
Clinical Stages of Classic Rett Syndrome
Classic Rett syndrome unfolds across four recognizable clinical stages. Knowing these stages is crucial for parents, because the transition from Stage 1 to Stage 2 — the terrifying period of rapid regression — is often misinterpreted as autism, cerebral palsy, or a parenting problem before the correct diagnosis is made.
Stage 1: Early Onset Stagnation (Ages 6–18 Months)
In the first six to twelve months of life, girls with Rett syndrome appear to develop normally — they smile, babble, show social interest, and reach early motor milestones. Then, subtly at first, development stalls. The infant may seem less engaged, make less eye contact, or show decreased interest in toys. Head growth decelerates, leading to acquired microcephaly — a head circumference that was normal at birth but falls below the normal range over time as the brain fails to grow at the expected rate.
These early changes are easily missed or attributed to temperament. Parents often describe a vague sense that something is "different" but are reassured at well-child visits because no clear regression has yet occurred. Stage 1 may last from a few months to a year, and in retrospect it is often recognized only after the more dramatic Stage 2 begins.
Stage 2: Rapid Destructive Phase (Ages 1–4 Years)
Stage 2 is the defining period of Rett syndrome, and for families it is the most devastating. Over weeks to months, previously acquired skills disappear:
- Loss of purposeful hand use: The ability to reach for objects, pick up food, or hold a toy — skills the child had genuinely mastered — vanish. This is a true regression, not a developmental delay. Parents describe their daughter putting something down one day and never picking it up again.
- Hand stereotypies: As purposeful hand function disappears, repetitive, involuntary hand movements emerge and become constant. The classic movement is a hand-wringing or hand-washing motion — the hands clasp together at the midline and move in a continuous washing, squeezing, or wringing pattern. These stereotypies are pathognomonic of Rett syndrome (present in essentially all cases) and persist throughout the child's life. The hands may also be brought to the mouth repeatedly.
- Language loss: First words, then babbling, then any intentional vocalization disappear. Girls who had said "mama" or "dada" lose these words. Some lose language so rapidly that parents describe it as happening "overnight."
- Autistic features: Social withdrawal, reduced eye contact, and apparent disengagement emerge during this period, frequently leading to an initial diagnosis of autism. However, unlike classic autism, these social features partially improve in Stage 3.
- Seizures: Epilepsy begins in 60–80% of girls during Stage 2, usually between ages 2 and 5. Seizure types vary: generalized tonic-clonic, myoclonic, and absence seizures are all seen.
- Abnormal breathing patterns: Waking-state breathing irregularities are a hallmark of Rett syndrome. Girls alternate between episodes of hyperventilation (rapid, deep breathing) and breath-holding (apnea), sometimes turning blue during the apneic episodes. Crucially, these breathing abnormalities occur only while awake — during sleep, breathing normalizes. This awake-only pattern distinguishes RTT breathing dysregulation from sleep apnea or central respiratory failure.
Stage 3: Plateau or Pseudo-Stationary Phase (Ages 2–10 Years)
After the turmoil of Stage 2, many girls enter a prolonged period of relative stability. Motor skills plateau rather than continuing to decline. Seizures often become somewhat better controlled with medication. Perhaps most movingly, girls in Stage 3 frequently show improvements in social engagement — they make better eye contact, seem more present, and may show flickers of improved communication. Many families describe Stage 3 as a period when they "get their daughter back" in some meaningful way, even though speech does not return. Hand stereotypies persist but may be less intense. Stage 3 is often the longest phase, lasting years to decades.
Stage 4: Late Motor Deterioration (Adult)
Stage 4 is characterized by progressive motor decline in older girls and women. Key features include:
- Scoliosis: Spinal curvature develops in the majority of patients and can be severe and rapidly progressive. Many require surgical spinal fusion. Managing scoliosis in RTT is complicated by the heightened anesthetic risk these patients carry (see Cardiac section).
- Parkinsonism-like features: Muscular rigidity and slow, effortful movement (bradykinesia) develop in many adults with RTT, resembling Parkinson disease features.
- Loss of ambulation: Some women who could walk in Stage 3 lose the ability to do so in Stage 4, though many retain ambulation into adulthood with consistent physical therapy.
- Cachexia: Progressive weight loss despite seemingly adequate caloric intake is a feature of Stage 4. The mechanism is not fully understood but involves autonomic dysfunction, increased energy expenditure from constant movement, and possible gastrointestinal dysmotility.
Women with Rett syndrome can live into their 50s and 60s. Quality of life and longevity are significantly influenced by the quality of supportive care, scoliosis management, and seizure control.
Diagnosis
Rett syndrome is primarily a clinical diagnosis — a physician who recognizes the characteristic pattern of normal early development followed by regression, loss of hand use, and emergence of hand stereotypies in a girl can diagnose RTT on clinical grounds alone. Genetic confirmation is then sought to solidify the diagnosis and exclude differential diagnoses.
Diagnostic Criteria
The revised diagnostic criteria published in 2010 in Annals of Neurology define two forms:
Classic (typical) Rett syndrome requires all four of the following:
- A period of regression followed by stabilization or partial recovery
- Loss of purposeful hand movements
- Loss of spoken language
- Emergence of repetitive, stereotyped hand movements
Atypical (variant) Rett syndrome requires at least two of the four main criteria plus at least five of eleven supportive criteria (including onset before age 5, scoliosis, breathing dysregulation, EEG abnormalities, and others). Atypical forms may have earlier or later onset, preserve some hand use, or have milder language loss.
Genetic Testing
MECP2 sequencing detects mutations in more than 95% of classic RTT cases. The eight most common mutations account for the majority of positive results. When sequencing is negative but clinical suspicion is high, deletion/duplication analysis should follow, as large deletions or duplications in MECP2 can cause RTT and are missed by sequencing alone.
It is important not to confuse classic RTT-causing mutations with MECP2 duplication syndrome, a completely separate condition. Males who carry an extra copy (duplication) of the entire MECP2 region develop a severe progressive encephalopathy with spastic quadriplegia, recurrent respiratory infections, and early death — a phenotype entirely distinct from classic Rett syndrome.
Variant Forms: CDKL5 and FOXG1
Two genes cause RTT-like phenotypes that were previously classified as atypical Rett syndrome but are now recognized as distinct conditions:
- CDKL5 deficiency disorder: Mutations in CDKL5 (cyclin-dependent kinase-like 5) cause seizures that begin in the first weeks of life, followed by RTT-like features including hand stereotypies and intellectual disability. The very early seizure onset (before age 5 months in most cases) distinguishes CDKL5 from classic RTT.
- FOXG1 syndrome (congenital variant): Mutations in the FOXG1 transcription factor gene cause a congenital form with profound intellectual disability, dyskinesia, and minimal or absent developmental milestone achievement from birth. These children never have a period of normal development, distinguishing them from classic RTT.
Differential Diagnosis
Before the characteristic hand stereotypies emerge and regression becomes obvious, Rett syndrome is most commonly mistaken for autism spectrum disorder (particularly in Stage 2 when autistic features are prominent) and cerebral palsy (due to motor difficulties). Angelman syndrome and other chromosomal disorders are sometimes considered in the differential. The combination of purposeful hand loss with emergence of midline hand-wringing movements in a previously developing girl is highly specific for RTT and should prompt immediate genetic testing.
Seizures and Breathing Irregularities
Epilepsy and breathing dysregulation are among the most challenging and frightening features of Rett syndrome for families. Understanding what is — and is not — dangerous is crucial for day-to-day management.
Epilepsy in RTT
Between 60% and 80% of girls with Rett syndrome develop epilepsy, most commonly during Stage 2. Seizures can take many forms: generalized tonic-clonic (the classic "grand mal"), myoclonic jerks, absence spells, and partial seizures with secondary generalization are all seen. EEG typically shows generalized spike-wave discharges, often with a characteristic slow background rhythm.
Seizure control is variable. Some girls respond well to antiepileptic medications and achieve good seizure control; others have refractory epilepsy that is difficult to manage. The most commonly used medications include:
- Lamotrigine: Often the first-line agent; generally well tolerated and effective for both tonic-clonic and absence seizures in RTT.
- Levetiracetam: Broad-spectrum agent with minimal drug interactions; may worsen behavioral irritability in some girls.
- Valproate: Effective for multiple seizure types; requires monitoring for hepatotoxicity and thrombocytopenia, and carries weight gain risk that complicates nutritional management in RTT.
- Carbamazepine: Sometimes used, particularly when breathing irregularities overlap with seizure activity.
Importantly, seizure frequency often decreases naturally in Stage 3, even without medication changes. What families and caregivers sometimes label as "seizures" in older girls may actually be non-epileptic events — movement stereotypies, autonomic episodes, or startle reactions — and distinguishing these from true ictal events requires video-EEG monitoring.
Breathing Irregularities: What They Are and Why They Happen
The breathing pattern abnormalities in Rett syndrome are one of the most distinctive and alarming features for caregivers. Girls alternate between episodes of hyperventilation (rapid, deep over-breathing), irregular breathing, and complete breath-holding (apnea) that can last seconds to over a minute. During apneic episodes, girls may turn blue (cyanotic), stiffen, or appear to lose consciousness briefly.
The critical reassurance for families: these breathing episodes occur only while awake. During sleep, breathing in RTT normalizes completely. This awake-only pattern is pathognomonic — it reflects dysfunction in the brain stem circuits that regulate voluntary breathing, not a structural airway problem or central respiratory failure. Girls are not at risk of dying in their sleep from these episodes.
Despite being frightening to witness, the breath-holding episodes, while capable of causing transient oxygen desaturation, rarely cause sustained harm. The brain's automatic breathing reflex eventually overrides the voluntary breath-hold and breathing resumes. There is currently no consistently effective pharmacologic treatment for the breathing irregularities in RTT, though carbamazepine has been tried with variable results. Management is primarily reassurance, education, and ensuring caregivers know to remain calm during episodes.
Cardiac Risks and Monitoring
Heart involvement in Rett syndrome is one of the most medically serious and underappreciated aspects of the condition. Women and girls with RTT have a substantially elevated risk of sudden unexpected death — and a major contributor to this risk is cardiac arrhythmia driven by prolonged QT interval.
Prolonged QT Interval in RTT
The QT interval on an electrocardiogram (ECG) represents the time it takes for the heart's ventricles to electrically recover after each heartbeat. When this interval is prolonged, the heart is vulnerable to a dangerous arrhythmia called torsades de pointes — a rapid, disorganized rhythm that can degenerate into ventricular fibrillation and sudden cardiac death.
Studies have found QT prolongation in approximately 40% of individuals with Rett syndrome. This is a far higher prevalence than the general population and reflects the role MeCP2 plays in regulating cardiac ion channel gene expression — particularly genes encoding the potassium channels that determine ventricular repolarization time.
Clinical Implications
The cardiac implications for RTT management are practical and important:
- Annual ECG screening is recommended for all RTT patients to track QTc interval length over time.
- QT-prolonging medications must be avoided. Many commonly used drugs — including certain antiepileptics, antipsychotics, antibiotics (azithromycin, clarithromycin, fluoroquinolones), and antihistamines — prolong the QT interval and can trigger fatal arrhythmias in susceptible individuals. Before prescribing any new medication to a patient with RTT, the prescriber must check the drug's cardiac profile.
- Anesthesia requires cardiac monitoring. The combination of QT prolongation and autonomic dysregulation makes general anesthesia significantly higher risk in RTT than in the general pediatric population. Scoliosis surgery — often necessary in RTT — therefore requires experienced cardiac monitoring and anesthesia teams familiar with the RTT cardiac phenotype.
- Sudden unexplained death in RTT (SUDEP-equivalent): Sudden death occurs in approximately 1–2% of RTT patients per year, a rate substantially higher than age-matched peers. Arrhythmia is thought to be a leading mechanism, though autonomic dysregulation contributing to respiratory arrest during an autonomous event may also play a role.
Families should be educated about cardiac risk and should carry a medication safety card listing drugs to avoid. Cardiologists familiar with RTT should be part of the multidisciplinary care team for affected individuals.
Management and Supportive Care
There is currently no cure for Rett syndrome. Management is multidisciplinary, supportive, and symptom-directed. Despite the severity of the condition, quality of life can be substantially improved with comprehensive care, and many women with RTT live meaningful lives with strong family and community support.
Seizure Management
Antiepileptic drug selection should be guided by seizure type, EEG findings, and the critical constraint of cardiac safety (avoiding QT-prolonging agents). Polypharmacy is common in refractory cases. The ketogenic diet has been used with some success in RTT-associated epilepsy when medications fail, and its high-fat composition may also benefit the nutritional challenges of Stage 4. Video-EEG monitoring helps distinguish true seizures from non-epileptic events, preventing unnecessary medication escalation.
Communication and Augmentative Technology
Perhaps the most important rehabilitation insight in RTT is that language comprehension is substantially preserved even when expressive language is completely absent. Girls with RTT understand far more than they can express. Eye-gaze communication technology — devices that track eye movements to select letters, words, or symbols on a screen — has been transformative for many families, allowing affected individuals to express preferences, answer questions, and participate in decisions about their own care. Low-tech options (picture boards, eye-pointing to choices) provide access when electronic devices are unavailable. Speech-language pathologists specializing in augmentative and alternative communication (AAC) are essential members of the RTT care team.
Physical and Occupational Therapy
Physical therapy focuses on maintaining mobility, weight-bearing, and muscle tone. Walking ability in Stage 3 is associated with better long-term motor outcomes, and structured PT programs help preserve ambulation. Aquatic therapy is particularly well-tolerated in RTT and may improve tone regulation, balance, and mood. Occupational therapy addresses hand use, adaptive equipment, and daily living skills, working around the involuntary hand stereotypies.
Scoliosis Management
Scoliosis develops in the majority of RTT patients and requires active monitoring with spine radiographs at regular intervals. Mild curvature may be managed with positioning and supportive seating. Progressive curvature is initially approached with bracing, though bracing is less effective in RTT than in idiopathic adolescent scoliosis. Severe curvature (generally greater than 40–50 degrees) typically requires surgical spinal fusion. Surgical planning must account for the elevated cardiac and anesthetic risks described above, and families should be referred to surgical teams experienced with neurologically complex patients.
Nutritional Support
Weight management in RTT is complicated by two opposing forces: the increased caloric expenditure from constant movement and stereotypies, and the progressive dysphagia and chewing difficulties that reduce intake in Stage 4. High-calorie dietary supplementation is frequently needed. Gastrostomy tube (G-tube) placement provides reliable nutritional access for patients with severe dysphagia or weight loss, and many families report improved quality of life after G-tube placement because mealtimes become less stressful and nutrition becomes more reliable.
Autonomic and Sleep Dysfunction
Autonomic nervous system dysregulation underlies many RTT symptoms beyond breathing: constipation, cold hands and feet, irregular heart rate variability, and temperature regulation problems are common. Sleep disturbances — difficulty falling asleep, frequent night waking, and daytime sleepiness — affect the majority of RTT patients and significantly impact family functioning. Melatonin is the most commonly used sleep aid, with reasonable safety data in this population. Environmental modifications (consistent schedules, darkening, white noise) are complementary.
Trofinetide (DAYBUE) — FDA-Approved 2023
In March 2023, the U.S. Food and Drug Administration approved trofinetide (brand name DAYBUE) for the treatment of Rett syndrome in patients aged 2 years and older — making it the first drug ever specifically approved for RTT. This approval marked a historic milestone after decades in which management was entirely symptomatic and no drug had demonstrated efficacy in rigorous clinical trials.
What Is Trofinetide?
Trofinetide is a synthetic analog of glycine-proline-glutamate (GPE), a naturally occurring tripeptide derived from insulin-like growth factor 1 (IGF-1). It acts through two complementary mechanisms relevant to RTT pathophysiology: it modulates IGF-1 receptor signaling, which supports neuronal survival and synaptic plasticity, and it reduces neuroinflammation by suppressing microglial activation. Both pathways are implicated in the synaptic deficits observed in RTT brains.
The LAVENDER Trial
The approval was based largely on the Phase 3 LAVENDER trial, published in The Lancet Neurology in 2023. This randomized, double-blind, placebo-controlled trial enrolled 187 girls with RTT aged 5 to 20 years. Patients receiving trofinetide showed statistically significant improvements compared to placebo on the Rett Syndrome Behaviour Questionnaire (RSBQ) and a caregiver-reported global impression of change scale. Improvements were observed in communication, purposeful hand use, and breathing patterns — the core deficits of RTT.
What to Expect from Trofinetide
Trofinetide is not a cure, and it does not reverse the underlying genetic defect. Families should understand that the improvements seen in trials, while statistically significant and clinically meaningful to affected individuals, are modest in absolute terms. Not every patient responds, and the degree of benefit varies. However, for a disease with no prior pharmacologic treatment, even modest gains in communication or reduction in distressing behaviors represent a meaningful advance.
Trofinetide is administered orally as a liquid solution, dosed by weight twice daily. The most common side effects are gastrointestinal: diarrhea occurs in approximately 80% of patients and weight loss in a substantial proportion. These side effects led to discontinuation in a minority of trial participants. Dose reduction and supportive management (anti-diarrheal agents, dietary adjustments) can help manage GI effects. The drug is expensive, and insurance coverage requires prior authorization with documentation of RTT diagnosis.
Earlier Trials
A smaller Phase 2 double-blind randomized trial published in Pediatric Neurology in 2019 first demonstrated trofinetide's safety profile and provided preliminary efficacy signals that supported the larger LAVENDER trial design. The progression from a promising Phase 2 result to FDA approval took approximately four years, an exceptionally rapid timeline for a rare pediatric neurological condition.
Gene Therapy and Future Directions
The identification of a single causative gene — MECP2 — made Rett syndrome an early and natural target for gene therapy approaches. The proof-of-concept was established in a landmark 2007 Science paper by Justine Guy and Adrian Bird at the University of Edinburgh, who showed that reactivating MeCP2 expression in adult mice with established RTT-like neurological symptoms reversed those symptoms. This experiment demonstrated that RTT is not simply a developmental catastrophe — mature neurons retain the capacity to recover if MeCP2 function is restored. This finding fundamentally changed the research landscape and launched a wave of gene therapy development.
The Challenge: MeCP2 Dosage Is Critical
Gene therapy for RTT faces a fundamental and unusual challenge: MeCP2 expression must be precisely calibrated. Too little MeCP2 causes RTT; too much MeCP2 is equally toxic. Males who carry a duplication of the MECP2 gene — producing about twice the normal amount of MeCP2 protein — develop a severe progressive encephalopathy called MECP2 duplication syndrome. This means gene therapy cannot simply deliver as much MeCP2 as possible; it must deliver the right amount to each neuron. Achieving this precision in vivo, across billions of neurons in a human brain, is a formidable technical problem.
Current Clinical Trials
Several gene therapy approaches are in active Phase 1/2 clinical development:
- AAV-MECP2 intrathecal delivery: Adeno-associated viral vectors carrying a functional MECP2 gene are injected into the spinal fluid (intrathecal space), allowing widespread distribution to neurons throughout the brain and spinal cord. Regulatory elements in the vector's promoter region are designed to limit MeCP2 expression to levels that mirror natural expression, preventing over-expression toxicity.
- Self-complementary AAV approaches: Modifications to standard AAV vectors designed to improve transduction efficiency in neurons without increasing dosing.
- RNA-based strategies: Antisense oligonucleotides and other RNA-targeting approaches aimed at modulating MeCP2 expression levels rather than replacing the gene outright.
Early results from Phase 1 trials are being reported, with initial safety data appearing acceptable. Efficacy signals are being evaluated carefully, with the caveat that Phase 1 trials are primarily designed to establish safe dosing, not to demonstrate efficacy.
Other Therapeutic Targets
Beyond gene therapy, several other biological targets are under investigation:
- IGF-1 and related pathways: Recombinant IGF-1 (mecasermin) has been studied in RTT; results have been modest but the pathway remains a therapeutic target, which is part of the rationale for trofinetide.
- BDNF enhancement: Brain-derived neurotrophic factor is reduced in RTT brains; strategies to increase BDNF signaling are under investigation.
- Serotonergic agents: Given the serotonin pathway disruption in RTT, agents modulating serotonin signaling (including dextromethorphan and others) have been explored with limited results to date.
- X-inactivation manipulation: Targeted efforts to preferentially silence the mutant X chromosome in more cells — effectively increasing the proportion of neurons producing normal MeCP2 — represent an elegant conceptual approach, though implementation in differentiated neurons remains technically challenging.
The RTT research community has been energized by the success of trofinetide's approval and by the promise of gene therapy trials. For a condition that had no disease-modifying treatments for decades, the current pipeline represents unprecedented momentum.
Key Research Papers
- Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. PMID 10508514 — The landmark paper identifying MECP2 mutations as the cause of Rett syndrome.
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471–479. PMID 6638958 — The first major English-language clinical description that established RTT as a recognized syndrome.
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27(3):322–326. PMID 11242117 — Established the Mecp2-null mouse as the definitive animal model for RTT research.
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143–1147. — Search PubMed — Demonstrated that restoring MeCP2 expression in adult symptomatic mice reversed RTT-like symptoms, opening the door to gene therapy.
- Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944–950. — Search PubMed — The current consensus diagnostic criteria used clinically worldwide.
- Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51(3):152–158. — Search PubMed — Genotype-phenotype analysis characterizing which mutations associate with milder versus more severe clinical outcomes.
- Glaze DG, Neul JL, Percy A, et al. A double-blind, randomized, placebo-controlled clinical study of trofinetide in the treatment of Rett syndrome. Pediatr Neurol. 2019;95:23–29. — Search PubMed — Phase 2 clinical trial establishing trofinetide's safety profile and preliminary efficacy signals.
- Neul JL, Percy AK, Benke TA, et al. Trofinetide for Rett syndrome (LAVENDER trial): a randomized, double-blind, placebo-controlled phase 3 study. Lancet Neurol. 2023;22(9):773–784. PMID 37507155 — The pivotal Phase 3 trial supporting FDA approval of trofinetide (DAYBUE) in 2023.
- Weaving LS, Ellaway CJ, Gecz J, Christodoulou J. Rett syndrome: clinical review and genetic update. J Med Genet. 2005;42(1):1–7. — Search PubMed — Comprehensive clinical review covering molecular genetics, phenotypic spectrum, and differential diagnosis.
- Moser SJ, Weber P, Lutschg J. Rett syndrome: clinical and electrophysiologic aspects. Pediatr Neurol. 2007;36(2):95–100. — Search PubMed — Characterizes the cardiac QTc prolongation and electrophysiologic features relevant to sudden death risk in RTT.
- Katz DM, Bird A, Coenraads M, et al. Rett syndrome: crossing the threshold to clinical translation. Trends Neurosci. 2016;39(2):100–113. — Search PubMed — Reviews the translational pipeline from mouse models to clinical trials and identifies key remaining hurdles.
- Ip JPK, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018;19(6):368–382. — Search PubMed — State-of-the-art review of MeCP2 biology, circuit-level dysfunction, and therapeutic targets.
PubMed Topic Searches
- Rett syndrome MECP2 gene therapy — PubMed
- Rett syndrome trofinetide clinical trial — PubMed
- MECP2 epigenetic neurodevelopmental — PubMed
- Rett syndrome QT prolongation cardiac — PubMed
- Rett syndrome X-inactivation mosaic — PubMed
Connections
- Genetics
- Angelman Syndrome — the closest differential diagnosis: another neurodevelopmental disorder with absent speech, seizures, and stereotyped hand movements.
- Autism Spectrum Disorder (ASD) — the autistic features of Stage 2 regression frequently lead to an initial autism diagnosis before RTT is recognized.
- Epilepsy — 60–80% of girls with Rett syndrome develop seizures, usually during the Stage 2 regression period.
- Long QT Syndrome — QT prolongation affects roughly 40% of RTT patients and drives the elevated sudden-death and anesthetic risk.
- Scoliosis — spinal curvature develops in the majority of RTT patients and often requires bracing or surgical fusion.