Prader-Willi Syndrome
- Overview and Genetics
- Genomic Imprinting and Chromosome 15q11-13
- Molecular Mechanisms: Deletion, UPD, and Imprinting Defect
- Neonatal and Infant Presentation
- Childhood and Adult Features: Hyperphagia and Obesity
- Endocrine Features: Growth Hormone and Hypogonadism
- Behavioral and Neurodevelopmental Features
- Diagnosis
- Treatment and Management
- Key Research Papers
- Connections
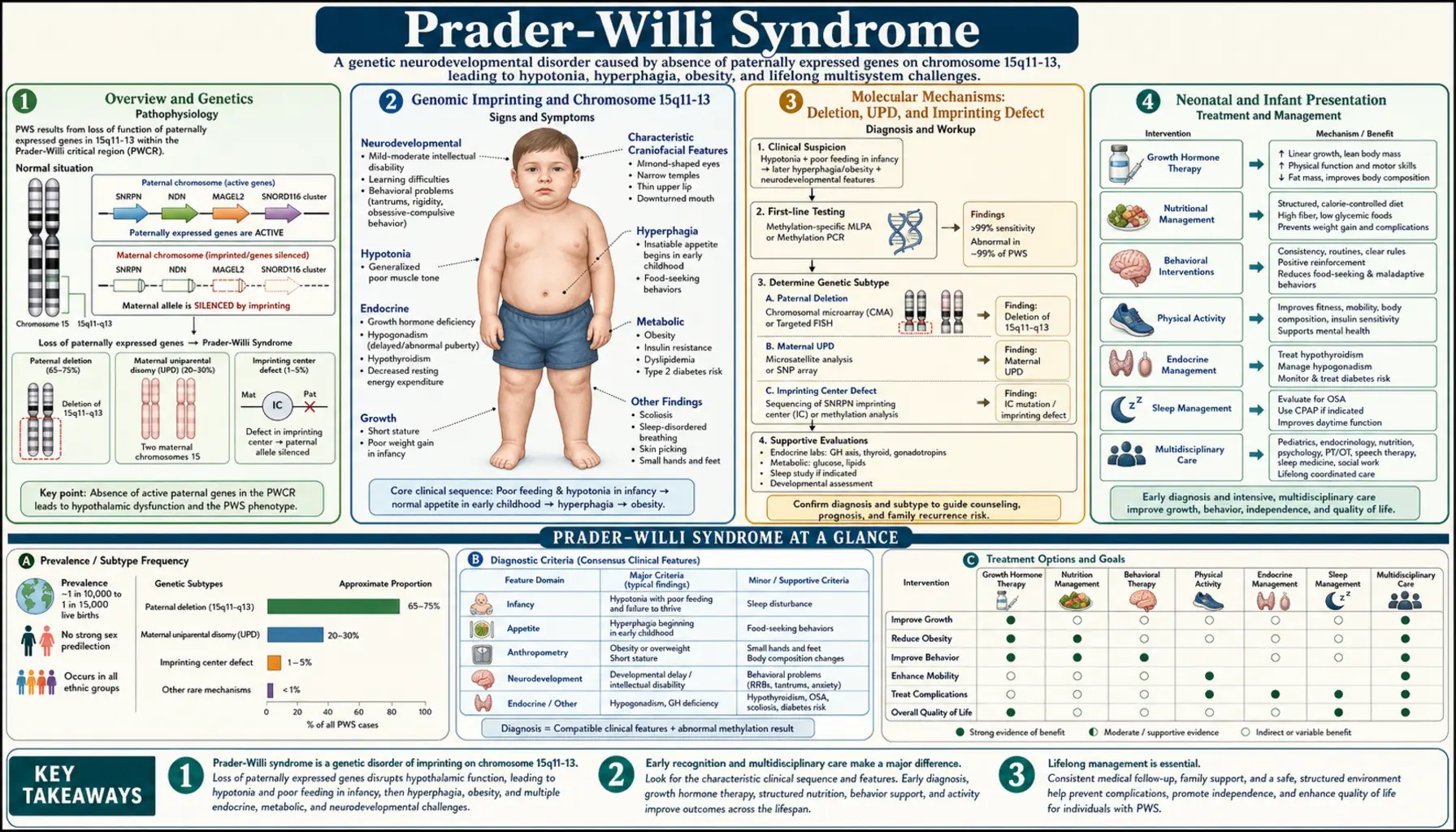
Overview and Genetics
Prader-Willi syndrome (PWS) is a rare but serious genetic disorder caused by the absence of functioning copies of paternally expressed genes on a specific region of chromosome 15. It affects approximately 1 in 15,000 to 25,000 live births, making it uncommon but not exceptionally rare — several thousand people are born with PWS every year in the United States alone. PWS holds the distinction of being the most common genetic cause of life-threatening obesity and ranks among the most frequently identified syndromic causes of intellectual disability worldwide. Despite its rarity, it is one of the best-studied genomic imprinting disorders and has taught researchers a tremendous amount about how genes are selectively silenced based on which parent they come from.
The syndrome was first described in 1956 by Swiss physicians Andrea Prader, Alexis Labhart, and Heinrich Willi, who noted a cluster of features in nine children: extreme muscle weakness at birth, followed later by insatiable appetite, obesity, intellectual disability, and small stature. For decades, the underlying cause remained unknown. It was not until the 1980s and 1990s that advances in cytogenetics and molecular biology revealed that PWS results from a failure of the paternal copy of chromosome 15q11-13 to contribute its normal gene products to the developing brain. This discovery was groundbreaking not just for PWS patients, but for the entire field of human genetics, because it demonstrated for the first time that the same chromosomal region could cause two completely different diseases depending on which parent's copy was affected.
PWS affects virtually every organ system and every stage of life, but it does so in strikingly different ways at different ages. In the newborn period, the disorder presents primarily as profound muscle weakness (hypotonia) and feeding failure. During early childhood, the picture reverses dramatically: muscle tone improves but an insatiable, neurologically driven hunger emerges that, without strict environmental controls, leads to severe obesity and life-threatening complications. Managing PWS is a lifelong, multidisciplinary effort involving pediatric endocrinologists, neurologists, behavioral therapists, dietitians, and family caregivers working in close coordination.
Genomic Imprinting and Chromosome 15q11-13
To understand PWS, it helps to understand genomic imprinting — one of the most fascinating and counterintuitive phenomena in all of genetics. Normally, every person inherits two copies of each chromosome, one from their mother and one from their father, and in most cases both copies are active and contribute equally to gene expression. Genomic imprinting is the exception to this rule: for a small subset of genes scattered throughout the genome, only one copy is active, and which copy is active is determined entirely by which parent it was inherited from. This "parental stamp" is not written in the DNA sequence itself but in chemical marks (primarily DNA methylation and histone modifications) placed on the chromosomes during the formation of eggs and sperm. These marks are reset each generation — a woman's eggs carry the maternal imprint regardless of whether her own father or mother contributed each chromosome to her.
Chromosome 15q11-13 is one of the most important imprinted regions in the human genome. This stretch of DNA contains a cluster of genes that are only active on the chromosome inherited from the father, along with other genes that behave oppositely. The paternally expressed genes in this region include SNRPN (which encodes a protein involved in RNA processing), MAGEL2 (linked to circadian rhythms and muscle function), NDN (involved in neuronal survival), MKRN3 (a regulator of puberty onset), and most importantly, a cluster of small nucleolar RNAs called SNORD116 (also known as PWCR1). The SNORD116 cluster is now believed to be the critical driver of most PWS features, particularly the abnormal hunger regulation, because of its specific expression in neurons of the hypothalamus — the brain region that controls appetite, metabolism, and hormone release. The maternal copy of this region has these genes chemically silenced (turned off) in every cell in the body.
The same chromosomal region also contains genes that work in the opposite direction: UBE3A, which encodes an enzyme (ubiquitin ligase) involved in protein degradation, is expressed only from the maternal copy in neurons. This single detail explains why the same chromosomal region causes two entirely different syndromes. In PWS, the paternal contribution is missing, so the brain loses all of the paternally expressed genes (SNRPN, MAGEL2, NDN, MKRN3, SNORD116). In Angelman syndrome, the maternal contribution is missing, so neurons lose UBE3A — the only imprinted gene in the region whose maternal copy is the active one in brain cells. The result is completely different: PWS causes hypotonia, hyperphagia, and mild-to-moderate intellectual disability; Angelman syndrome causes severe intellectual disability, absent speech, seizures, and a distinctive happy affect. Same location, same size of DNA change, opposite parent of origin — opposite diseases. This contrast remains one of the clearest demonstrations that in genetics, where a gene comes from can matter as much as what the gene is.
Molecular Mechanisms: Deletion, UPD, and Imprinting Defect
PWS arises from three distinct molecular mechanisms, all of which produce the same end result: the brain lacks functioning copies of the paternally expressed genes at 15q11-13. The most common mechanism, accounting for roughly 70% of cases, is a paternal deletion — a de novo (new, not inherited) deletion of approximately 5 to 6 million base pairs on the paternally inherited chromosome 15. Because this region is already silent on the maternal copy due to imprinting, losing it from the paternal copy means the brain has no working copies at all. These deletions are detected by fluorescence in situ hybridization (FISH), chromosomal microarray, or multiplex ligation-dependent probe amplification (MLPA). The deletion is almost always new in each family, meaning parents who have one child with a deletion have only a slightly elevated (less than 1%) risk for future children compared to the general population.
The second mechanism, responsible for about 25% of cases, is maternal uniparental disomy (UPD). In UPD, both copies of chromosome 15 in the child are inherited from the mother, with no paternal copy present at all. Since the maternal copy of 15q11-13 is imprinted (silenced) for the paternal-specific genes, having two maternal copies is functionally equivalent to having no copies — the brain still cannot express SNORD116, MAGEL2, or the other paternally expressed genes. Maternal UPD usually arises from a process called trisomy rescue: an egg containing two copies of chromosome 15 (instead of the normal one) is fertilized by a normal sperm, creating a cell with three copies of chromosome 15. The cell then corrects this by losing one copy, and by chance, the lost copy may be the paternal one — leaving two maternal copies. Because this process requires an abnormal egg, the mother's age slightly increases the risk, though most UPD cases occur in younger mothers.
The remaining 5% of cases involve an imprinting defect — the chromosomal DNA is structurally intact, with one maternal and one paternal copy present, but the paternal copy has been given the wrong chemical marks. It carries maternal-type methylation patterns instead of paternal ones, so the brain silences the paternal genes just as it would silence the maternal ones. This can happen spontaneously (a random error in establishing imprints during sperm formation) or it can be inherited — a small percentage of imprinting defects are caused by inherited deletions within the imprinting center, a regulatory region that controls the imprinting of the entire domain. Families with inherited imprinting center deletions face much higher recurrence risks (up to 50% for certain inheritance patterns), making genetic counseling essential. There are subtle genotype-phenotype differences among the three mechanisms: patients with UPD tend to have milder hypotonia at birth and somewhat less severe feeding difficulties as infants, but they show more pronounced behavioral challenges, autism-spectrum features, and psychiatric vulnerability in later life. Deletion patients often display more intense food-seeking behavior and may have lighter skin and hair coloration relative to their family members, because the deleted region includes the OCA2 gene which influences pigmentation.
Neonatal and Infant Presentation
The most striking feature of PWS in newborns is profound, generalized hypotonia — a severe reduction in muscle tone that earns these babies the clinical description "floppy infant." The hypotonia is central in origin, meaning it comes from the brain rather than from the muscles or peripheral nerves themselves, reflecting the absence of critical genes in the hypothalamus and other brain regions. Affected newborns lie with their limbs extended and limp, have a weak or absent cry, show poor arousal, and suck ineffectively. Many require admission to a neonatal intensive care unit (NICU) due to respiratory difficulties: the weakness of breathing muscles combined with poor airway tone creates a genuine risk of aspiration and respiratory failure. Some families report that their PWS baby moved less frequently during pregnancy — reduced fetal movement can be an early clue in retrospect, though it is rarely recognized as such before birth.
Feeding is a major challenge throughout the first year of life. Because the suck-swallow reflex requires coordinated muscle activity, infants with PWS cannot sustain effective breastfeeding or bottle feeding. They tire quickly during feeds, fail to extract enough milk, and lose weight despite their parents' best efforts. Nasogastric (NG) tube feeding or gavage feeding is commonly required for weeks to months while the infant's strength gradually improves. Occupational therapists and feeding specialists become essential team members early on, working to maximize whatever oral feeding the infant can manage while supplementing by tube. Failure to thrive in the first year, despite the baby's apparent lack of appetite, is one of the most disorienting aspects of PWS for new families — they will soon learn that this is the calm before a very different storm.
Physical features in the newborn period include a narrow forehead (narrow bifrontal diameter), almond-shaped eyes that may appear to slant slightly downward at the outer corners, a thin upper lip, and corners of the mouth that turn downward, giving a somewhat sorrowful expression even at rest. Hands and feet are characteristically small. The genitalia are frequently underdeveloped: boys typically have an undescended testicle (cryptorchidism) in one or both sides, a small penis (micropenis), and a reduced scrotum; girls have small labia minora and clitoris. These genital findings reflect central hypogonadism — the hypothalamus is not producing enough gonadotropin-releasing hormone (GnRH) to stimulate normal genital development. In deletion-type PWS, the affected baby may also appear noticeably fairer in skin and hair color compared to other family members, because the deleted region removes one copy of the pigmentation gene OCA2.
Childhood and Adult Features: Hyperphagia and Obesity
Between the ages of roughly 2 and 5, a dramatic shift occurs in children with PWS. The hypotonia that dominated infancy gradually improves — children become stronger, start walking (typically with a delay of 1-2 years), and the feeding difficulties that required tube support recede. If families are not warned about what comes next, this period of apparent improvement can be misleading. What emerges in its place is one of the most challenging and medically dangerous features of any genetic syndrome: hyperphagia, an unrelenting, neurologically driven hunger that never goes away. Unlike the normal hunger that healthy people feel when they have not eaten for several hours, the hunger in PWS is constant and does not diminish meaningfully after meals. Children with PWS report feeling hungry all the time, and the hunger they describe goes beyond simple appetite — it has a compulsive, consuming quality that dominates their mental life.
The underlying mechanism centers on the hormone ghrelin. Ghrelin is sometimes called the "hunger hormone" because it is released by the stomach when it is empty, travels through the bloodstream to the hypothalamus, and signals that it is time to eat. In healthy people, ghrelin levels fall sharply after a meal as the stomach fills, removing the hunger signal. In PWS, this normal feedback loop is broken. Plasma ghrelin levels in PWS patients are extraordinarily elevated — among the highest measured in any human condition — and crucially, they do not fall after eating. A person with PWS feels the same intense biological hunger signal after a full meal as a healthy person feels after skipping dinner and most of breakfast. Research points to the SNORD116 snoRNA cluster, whose loss disrupts the hypothalamic circuits that normally coordinate ghrelin secretion and satiety signaling, as the likely driver of this dysregulation.
The behavioral consequences of this unrelenting hunger are profound. Children and adults with PWS engage in food-seeking behaviors that can seem extreme or even bizarre to those who do not understand the neurological basis. They rummage through trash cans, hoard food in their rooms, eat pet food, consume frozen or raw ingredients, and in severe cases eat non-food items (pica). They think about food almost constantly, talk about food obsessively, and experience intense anxiety and distress when meals are delayed or when food is visible but inaccessible. Caregivers who attempt to restrict food access — even with the best intentions — may encounter explosive tantrums, deception, and determined food-seeking that seems almost impossible to manage. This behavior is not willful defiance; it is the expression of a biological drive as powerful as suffocation-driven air-seeking, just directed at food.
Without strict environmental controls, morbid obesity develops rapidly and brings life-threatening complications. Type 2 diabetes, obstructive sleep apnea, right heart strain (cor pulmonale) from chronic hypoxia, and cardiorespiratory failure are major causes of disability and premature death in PWS. Studies suggest that the average life expectancy without modern management was significantly reduced, with obesity-related complications accounting for most excess mortality. This is why families living with PWS must implement what amounts to a medically supervised locked-food-access protocol: refrigerators and pantries with physical locks, structured meal schedules with calorie-controlled portions, and strict monitoring of any food access outside the home at school, restaurants, or family gatherings. This is not parental harshness — it is the medical equivalent of providing a wheelchair to someone who cannot walk. Environmental control is as essential a medical intervention as any pill or injection.
Endocrine Features: Growth Hormone and Hypogonadism
Growth hormone (GH) deficiency is nearly universal in PWS and contributes significantly to the disorder's physical features and health burden. The deficiency arises from dysfunction in the hypothalamus, which fails to produce adequate growth hormone-releasing hormone (GHRH) to drive normal GH secretion from the pituitary gland. The effects are substantial: children with untreated PWS typically reach adult heights of only 150 to 160 centimeters (roughly 4 feet 11 inches to 5 feet 3 inches), well below expected family height. Beyond short stature, GH deficiency in PWS worsens the already problematic body composition — GH is a major driver of lean muscle mass and helps keep body fat stores in check. Without it, fat accumulates more readily and muscle remains sparse, creating a body composition that makes physical activity harder and caloric burn lower. Bone mineral density is also reduced, raising the long-term risk of osteoporosis and fractures.
Recombinant human growth hormone therapy was approved by the FDA specifically for PWS in 2000, and it has become one of the most important treatments in the syndrome's management. When started in infancy — ideally by 3 to 6 months of age — GH therapy produces remarkable benefits that extend far beyond height. Children on GH show improved muscle tone and motor development, better physical strength and endurance, improved cognitive function and learning, and significantly better body composition with more lean mass and less fat. The improvement in muscle tone can reduce the severity of feeding difficulties in infancy, making earlier initiation valuable even before height velocity becomes a concern. GH therapy is continued through childhood and adolescence for growth benefits, and increasingly through adulthood as well, because the body composition and metabolic benefits persist regardless of age. Clinicians monitor treated patients closely for scoliosis (spinal curvature, which can be worsened by rapid growth) and sleep apnea (which can occasionally be triggered or worsened by GH-related tonsillar enlargement), but neither complication prevents GH use — they require monitoring and management alongside it.
Central hypogonadism is the other major endocrine feature of PWS. The hypothalamus produces insufficient GnRH, which means the pituitary gland does not receive the signal to release LH and FSH, and without those gonadotropins, the gonads do not produce adequate sex hormones. In boys, cryptorchidism (undescended testicles) is present in 80-90% of cases and often requires surgical correction (orchiopexy) before age 1 to 2 years to preserve testicular function and reduce the risk of malignancy. Puberty in PWS, if it occurs at all, is typically incomplete — boys may develop some pubic hair but fail to fully virilize, while girls may begin breast development but often do not menstruate. Adults of both sexes are generally infertile. Sex hormone replacement therapy is used to induce puberty at the appropriate developmental age, and adults require ongoing hormone replacement for bone density, cardiovascular health, and overall quality of life. Some evidence also suggests that early testosterone supplementation in infant boys improves genital development and may have behavioral benefits, though practice varies by center. Clinicians should also be alert to reports of blunted cortisol response to stress in some PWS patients; adrenal insufficiency-like states during illness or surgery have been described, and stress dosing protocols may be appropriate in clinical situations where the patient is acutely unwell.
Behavioral and Neurodevelopmental Features
The intellectual and behavioral features of PWS are nearly as defining as the metabolic ones. Most individuals with PWS have mild to moderate intellectual disability, with IQ scores typically in the range of 55 to 70. A minority score in the borderline intellectual functioning range (70-85), and a small number achieve IQ scores in the average range, particularly among those with the UPD subtype. Academic strengths often include reading decoding and visual processing, while weaknesses tend to cluster around working memory, mathematics, and sequential processing. Children with PWS generally do well in structured, supportive educational environments and benefit significantly from special education services that accommodate their processing style and behavioral needs.
The behavioral phenotype of PWS is distinctive and can be among the most challenging aspects of the disorder for families. Temper tantrums are common and can be severe, triggered not only by food-related situations but also by unexpected changes in routine, perceived unfairness, or demands that exceed the individual's cognitive flexibility. Rigidity and inflexibility are core features: people with PWS frequently insist on sameness in daily routines, become highly distressed by changes in plans, and develop obsessive-compulsive behaviors around ordering, symmetry, collecting, or repetitive questioning. Stubbornness is a frequently cited challenge — once a person with PWS has formed an expectation or intention, redirecting them requires patience, predictability, and well-practiced behavioral strategies. Skin picking (excoriation) is extremely common and can range from mild to severe; some individuals create wounds requiring medical treatment, and the behavior appears driven by a combination of sensory-seeking and anxiety that is difficult to suppress through willpower alone.
Sleep disturbances are nearly universal in PWS and encompass several distinct problems. Excessive daytime sleepiness affects the majority of patients and can be severe enough to interfere with school, work, and daily functioning — some patients fall asleep mid-sentence or mid-activity. Central sleep apnea (from hypothalamic dysregulation of breathing during sleep) and obstructive sleep apnea (from obesity and reduced muscle tone) both occur and require polysomnography for proper diagnosis and treatment. Some PWS patients show narcolepsy-like features including sleep onset REM periods and cataplexy-like episodes. The UPD subtype carries elevated risk for psychiatric episodes, particularly psychotic disorders that can emerge in adolescence or early adulthood, sometimes without warning. Depression and anxiety are common across all subtypes. Pain sensitivity is often reduced in PWS — patients may not react normally to injuries, infections, or abdominal emergencies — which means caregivers and medical teams must monitor carefully for signs of illness that the patient may not report accurately.
Diagnosis
The clinical diagnosis of PWS is suspected on the basis of a characteristic constellation of features that shift with age: severe neonatal hypotonia and feeding failure in infancy, followed by hyperphagia and obesity in childhood, together with intellectual disability and the distinctive behavioral phenotype. Clinical diagnostic criteria were formalized by Holm and colleagues in 1993 and have been updated since, but molecular genetic testing is required to confirm the diagnosis definitively. Given the high treatability of PWS with growth hormone starting as early as infancy, and the critical importance of implementing dietary control early, prompt laboratory confirmation is essential — every month without treatment and dietary precautions in a child with PWS represents preventable developmental and metabolic harm.
DNA methylation analysis of the 15q11-13 region is the single best first-line test and detects more than 99% of PWS cases regardless of the underlying molecular mechanism. The test examines the pattern of chemical methylation marks on the chromosome: in a normally imprinted region, the paternal copy has one methylation pattern and the maternal copy has another, producing a recognizable mixed signal. When the paternal contribution is missing (whether by deletion, UPD, or imprinting defect), only the maternal methylation pattern is present, and this abnormal result confirms PWS. Methylation-specific PCR and methylation-specific MLPA are the most commonly used formats. A positive methylation result confirms that the child has PWS without specifying which of the three mechanisms is responsible. Determining the mechanism requires a second round of testing: FISH or chromosomal microarray identifies a deletion by detecting reduced copy number at 15q11-13; if no deletion is found, microsatellite analysis (short tandem repeat testing) of the family trio (child, mother, father) can distinguish UPD from an imprinting defect by establishing whether both chromosome 15 copies in the child came from one parent. This distinction matters for genetic counseling and recurrence risk assessment.
The differential diagnosis changes with age. In the newborn period, other causes of severe neonatal hypotonia must be considered, including hypoxic-ischemic encephalopathy, spinal muscular atrophy, congenital myotonic dystrophy, and other neuromuscular disorders. In these contexts, the presence of cryptorchidism, the specific facial features, and the maternal history of reduced fetal movement together make PWS a leading consideration. In childhood, when hyperphagia and obesity dominate, other genetic obesity syndromes enter the differential: Bardet-Biedl syndrome (which also causes intellectual disability, obesity, and hypogonadism, but with retinal dystrophy and polydactyly as distinguishing features), Alstrom syndrome, and Cohen syndrome. Hypothyroidism should always be excluded as a treatable cause of hypotonia and obesity. The methylation test reliably distinguishes PWS from all of these alternatives when the clinical picture is consistent.
Treatment and Management
There is currently no cure for PWS, and the underlying genetic cause cannot be corrected with available therapies. However, a well-coordinated, proactive management plan dramatically improves life expectancy, body composition, cognitive development, and quality of life. The cornerstone of management is lifelong dietary supervision with strict environmental control of food access. This means physical locks on refrigerators, pantries, and anywhere else food is stored; structured, scheduled mealtimes with calorie-controlled, nutritionally complete portions; supervision during all food-accessible situations including school cafeterias, family events, and restaurants; and education of every adult in the child's life about the neurological basis of hyperphagia. Families who approach this as a behavioral problem rather than a medical one — who believe that better parenting, more willpower, or appropriate punishment can solve the food-seeking — tend to have children with more severe obesity and worse outcomes. Environmental control is not optional; it is the primary treatment for the metabolic aspects of PWS.
Growth hormone therapy, as described above, is started as early as possible — ideally in the first year of life. Subcutaneous GH injections (given daily, similar to insulin injections) are well tolerated by most patients and the benefits accumulate over time. The dose is weight-based and adjusted as the child grows. Side effects are generally mild and manageable, and the benefits across multiple domains — height, muscle mass, cognitive function, metabolism, bone density — justify its use even when obesity is already established. Sex hormone replacement therapy is given when the child reaches the expected age of puberty (typically age 11-12) if spontaneous puberty does not begin or is incomplete. Testosterone therapy in males and combined estrogen-progesterone in females is continued into adulthood as ongoing maintenance for bone and cardiovascular health. Orchiopexy for undescended testicles should be performed early, before age 2, rather than waiting.
Behavioral and psychiatric management is an equally essential pillar of care. Structured daily routines, predictable environments, and clear behavioral support plans help reduce tantrums and anxiety. Applied behavioral analysis (ABA) techniques are useful for managing rigidity and repetitive behaviors. SSRIs (selective serotonin reuptake inhibitors) are commonly used for the OCD-spectrum behaviors and anxiety, with variable but sometimes significant benefit. For skin picking, behavioral therapy combined with sensory strategies (occupational therapy), and sometimes low-dose SSRIs, can reduce the severity. Psychotic episodes in UPD patients are treated with low-dose antipsychotic medications, and monitoring for emerging psychiatric symptoms during adolescence is important. Sleep disorders are addressed with CPAP or BiPAP for apnea and with modafinil or stimulant medications for hypersomnia when it significantly impairs daily functioning.
Several investigational treatments are in active clinical trials and show genuine promise for changing the management of PWS. Intranasal oxytocin, a neuropeptide involved in social bonding and appetite regulation, has shown signals of benefit in small trials for reducing hyperphagia, improving social behavior, and decreasing skin picking. Carbetocin, a longer-acting oxytocin analog, is also in trials. GLP-1 receptor agonists — the same class as semaglutide (Ozempic/Wegovy) — are being studied specifically in PWS because they act on the same hypothalamic appetite circuits that are dysfunctional in the syndrome, and early data suggest meaningful reductions in hyperphagia and body weight. Ghrelin receptor antagonists, which would directly block the excessive hunger signal, represent a mechanistically compelling approach and are under preclinical and early clinical investigation. As understanding of the SNORD116 region's function in the hypothalamus deepens, gene-targeted approaches — including antisense oligonucleotides that might activate the silenced maternal copy of imprinted genes — are being explored as potential disease-modifying strategies, though these remain several years from clinical application. Families should be encouraged to connect with the Prader-Willi Syndrome Association (PWSA) USA and with academic centers specializing in PWS to access clinical trial opportunities and current best-practice management guidelines.
Key Research Papers
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10-26. — Search PubMed
- Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38(12):1249-1263. PMID 26062517
- Butler MG, Manzardo AM, Forster JL. Prader-Willi syndrome: clinical genetics and diagnostic aspects with treatment approaches. Curr Pediatr Rev. 2016;12(2):136-166. PMID 26592417
- Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. 2011;155A(5):1040-1049. PMID 21465655
- Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91(2):398-402. PMID 8424017
- Haqq AM, Muehlbauer MJ, Newgard CB, et al. Ghrelin, peptide YY, glucose-dependent insulinotropic polypeptide, and hunger in Prader-Willi syndrome. J Pediatr. 2011;159(1):31-36. — Search PubMed
- Dykens EM, Cassidy SB. Prader-Willi syndrome: genetic, behavioral and treatment issues. Child Adolesc Psychiatr Clin N Am. 1996;5(4):913-927. — Search PubMed
- Goldstone AP, Holland AJ, Hauffa BP, et al. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93(11):4183-4197. — Search PubMed
- Carrel AL, Myers SE, Whitman BY, Allen DB. Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study. J Clin Endocrinol Metab. 2002;87(4):1581-1585. — Search PubMed
- Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome. In: Adam MP, et al., eds. GeneReviews. Search PubMed
- Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7(14):1-20. — Search PubMed
- Crinò A, et al. Growth hormone secretagogue receptor gene mutations in patients with Prader-Willi syndrome and implications for ghrelin pathways. J Pediatr Endocrinol Metab. 2012;25(3-4):291-299. — Search PubMed
Connections
- Genetics
- Angelman Syndrome
- Down Syndrome
- Turner Syndrome
- Growth Hormone Deficiency
- Congenital Adrenal Hyperplasia
- Hypothyroidism
- Endocrinology
- Psychiatry