Down Syndrome
- Overview and Epidemiology
- Cytogenetics and Mechanisms
- Physical Features and Dysmorphology
- Congenital Heart Disease
- Cognitive Development and Intellectual Disability
- Medical Complications
- Alzheimer's Disease Connection
- Prenatal Screening and Diagnosis
- Management and Therapies
- Key Research Papers
- Connections
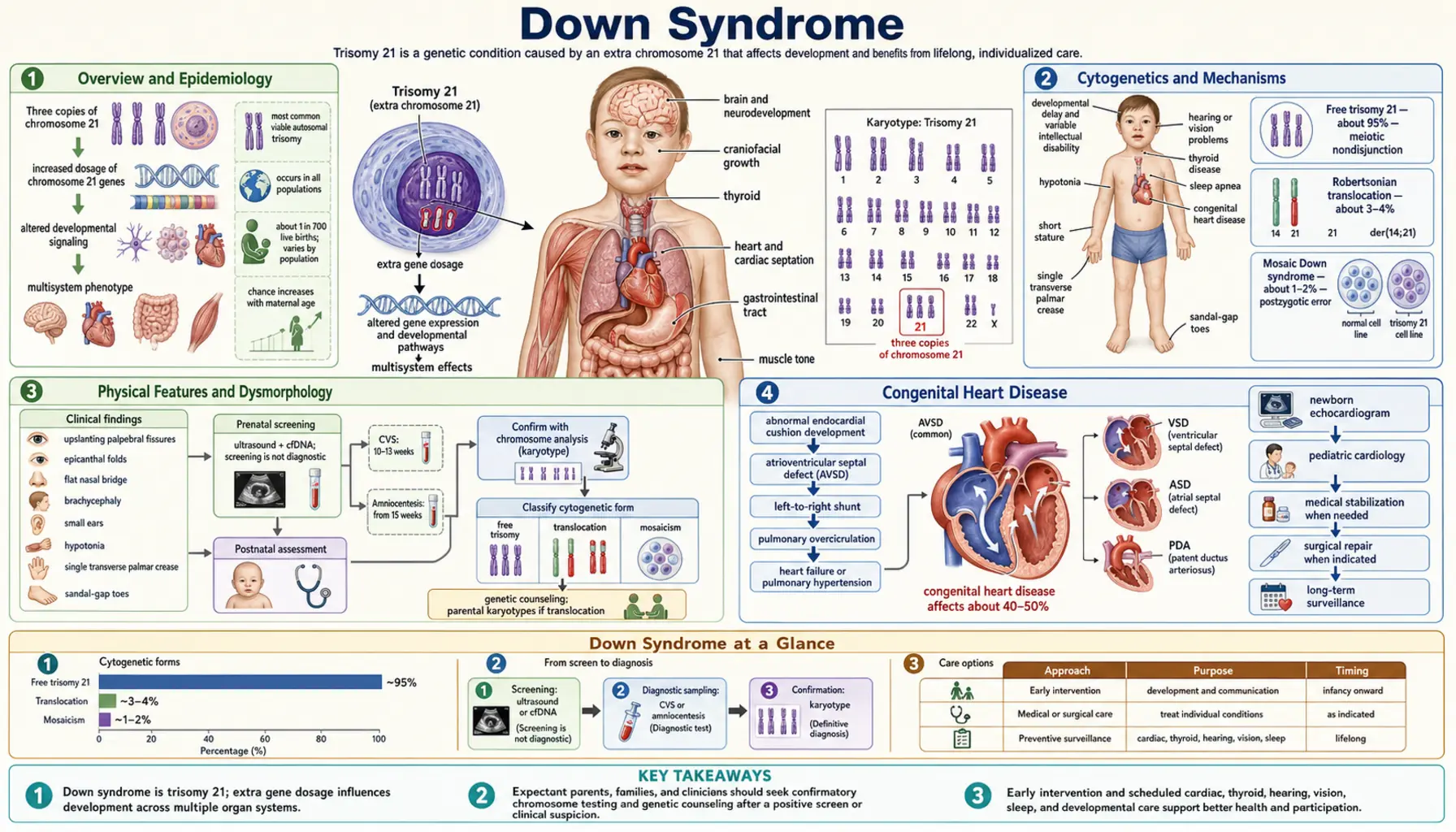
Overview and Epidemiology
Down syndrome, also known as Trisomy 21, is the most common chromosomal condition diagnosed in humans. It occurs in approximately 1 in every 700 live births, making it the leading chromosomal cause of intellectual disability worldwide. In the United States alone, roughly 6,000 infants are born with Down syndrome each year, and an estimated 200,000 Americans currently live with the condition.
The condition was formally described in 1866 by British physician John Langdon Down, who catalogued the distinctive physical features in a group of patients with intellectual disability. The chromosomal basis — an extra copy of chromosome 21 — was not established until 1959, when French geneticist Jérôme Lejeune identified trisomy 21 as the underlying cause.
Life expectancy has improved dramatically over the past six decades. In the 1960s, the median lifespan for a person with Down syndrome was approximately 10 years, primarily because congenital heart defects went untreated. Today, with modern cardiac surgery, comprehensive medical management, and early intervention programs, most individuals with Down syndrome live into their 60s or beyond. This dramatic improvement in longevity has shifted clinical focus toward adult health issues, including Alzheimer's disease and age-related conditions.
Down syndrome occurs across all racial, ethnic, and socioeconomic groups. While advanced maternal age is the strongest known risk factor, approximately 80% of children with Down syndrome are born to mothers under age 35, simply because younger women give birth far more frequently.
Cytogenetics and Mechanisms
Down syndrome results from the presence of three copies of chromosome 21 rather than the normal two. This extra genetic material — approximately 300–400 extra gene copies — alters gene dosage throughout development and produces the characteristic features of the syndrome. Three distinct cytogenetic mechanisms can cause this trisomic state.
Free trisomy 21 accounts for approximately 95% of cases. It arises from meiotic non-disjunction, the failure of chromosome 21 pair members to separate properly during gamete formation. In roughly 90% of free trisomy cases, the error occurs during maternal meiosis I. The resulting oocyte carries two copies of chromosome 21; when fertilized by a normal sperm, the embryo has three copies. The risk of non-disjunction rises steeply with maternal age: approximately 1 in 1,500 at age 20, 1 in 400 at age 35, and 1 in 25 at age 45. Recurrence risk for free trisomy 21 is roughly 1% above the age-related background risk.
Robertsonian translocation accounts for approximately 4% of cases. The long arm of chromosome 21 fuses to another acrocentric chromosome — most commonly chromosome 14, but also chromosomes 13 or 21. A carrier parent has only 45 chromosomes but a normal phenotype because no genetic material is lost. When one parent carries this balanced translocation, recurrence risk is substantially higher: 10–15% if the mother is the carrier, approximately 5% if the father is the carrier. Unlike free trisomy, translocation Down syndrome is not related to maternal age, making family genetic counseling essential.
Mosaic trisomy 21 accounts for approximately 1% of cases. It arises from a post-zygotic mitotic error after fertilization, producing a mixture of normal diploid cells and trisomic cells. The proportion of trisomic cells varies widely between individuals and between tissues. People with mosaic Down syndrome often — though not always — have a milder phenotype, and intellectual function is frequently higher than in full trisomy 21. The diagnosis can be missed on standard karyotyping if the sampling proportion is low.
A key gene on chromosome 21 relevant to several complications is APP (amyloid precursor protein), whose triple dosage drives accelerated amyloid-beta deposition and underlies the near-universal Alzheimer's pathology in adults with Down syndrome. DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A) is another trisomic gene implicated in cognitive deficits and is a current therapeutic target.
Physical Features and Dysmorphology
Down syndrome produces a recognizable constellation of physical features, though no single feature is present in every individual and severity varies considerably. The combination of findings, rather than any one sign, supports the diagnosis — which must always be confirmed by karyotype.
Hypotonia (low muscle tone) is virtually universal at birth and is often the first clinical clue. It contributes to feeding difficulties in infancy, delayed motor milestones, and the open-mouth posture seen in many young children. Tone generally improves with age and targeted physical therapy.
Characteristic craniofacial features include:
- Upslanting palpebral fissures — the outer corners of the eyes angle upward
- Epicanthal folds — skin folds at the inner corner of each eye
- Brushfield spots — speckled white or grey dots around the iris periphery, seen in approximately 90% of individuals with blue or grey eyes
- Brachycephaly — shortened anteroposterior head diameter with a flat occiput
- Flat nasal bridge and small, upturned nose
- Macroglossia — relatively large tongue that may protrude; contributes to articulation difficulties and obstructive sleep apnea
- Small, low-set ears, often with small ear canals predisposing to recurrent otitis media
- Short neck with excess skin at the nape in newborns
Limb and hand features include a single palmar crease (simian crease) in approximately 50% of individuals, a sandal gap (wide space between the first and second toes), and fifth finger clinodactyly (incurving of the little finger). Short stature is universal, with adult heights typically 20–30 cm below population norms.
Joint laxity is common and, combined with hypotonia, increases the risk of subluxation. Of particular clinical importance is atlantoaxial instability (C1–C2 subluxation), present in 10–15% of individuals. This carries a risk of spinal cord injury with high-impact activities or neck hyperextension during intubation, and is assessed by lateral cervical spine X-rays before sports participation or surgery.
Congenital Heart Disease
Congenital heart defects (CHD) are the most medically significant structural abnormality in Down syndrome, affecting 40–50% of all newborns with the condition. Before modern cardiac surgery became routine, CHD was the leading cause of early death. Today, with prompt diagnosis and repair, most individuals with Down syndrome and CHD achieve excellent cardiac outcomes.
The most common cardiac defect is atrioventricular septal defect (AVSD), also called AV canal defect or endocardial cushion defect, accounting for approximately 45% of CHD in Down syndrome. AVSD involves incomplete formation of the central cardiac skeleton, producing a combined atrial septal defect, ventricular septal defect, and abnormalities of the mitral and tricuspid valves. Down syndrome accounts for approximately 70% of all AVSD cases in the general population, reflecting the strong genetic predisposition.
Other common cardiac defects in Down syndrome include:
- Ventricular septal defect (VSD) — approximately 35% of CHD in DS
- Atrial septal defect (ASD) — approximately 8%
- Patent ductus arteriosus (PDA) — particularly in preterm births
- Tetralogy of Fallot — approximately 5% of CHD in DS
A critical concern with unrepaired left-to-right shunts (AVSD, large VSD, ASD) is Eisenmenger syndrome — irreversible pulmonary hypertension caused by prolonged excess pulmonary blood flow, which reverses the shunt direction and makes surgical repair no longer possible. Individuals with Down syndrome may develop Eisenmenger physiology more rapidly and at younger ages than the general population, making early detection and timely repair essential.
Because clinical examination alone is unreliable for detecting CHD in Down syndrome — particularly AVSD, which may not produce a murmur initially — echocardiography is recommended for all newborns with Down syndrome regardless of whether cardiac signs are present. This recommendation is explicit in current American Academy of Pediatrics guidelines.
Cognitive Development and Intellectual Disability
Intellectual disability (ID) is present in virtually all individuals with Down syndrome, but the degree varies substantially. IQ scores typically fall in the range of 35 to 70, corresponding to mild-to-moderate intellectual disability, with the majority clustering in the mild-to-moderate range (IQ 40–70). Individuals with mosaic Down syndrome may score higher. Importantly, IQ scores alone do not capture the full range of abilities and adaptive functioning, which can be considerably stronger than cognitive test scores suggest.
Cognitive strengths and weaknesses follow a characteristic profile. Visual-spatial processing and social cognition are relative strengths — many individuals with Down syndrome demonstrate strong face recognition, social awareness, and emotional intelligence. In contrast, auditory short-term memory and expressive language are areas of relative weakness. Vocabulary and receptive language typically exceed expressive language ability, meaning individuals often understand more than they can verbally express.
Early intervention — beginning in infancy — has the most robust evidence for improving long-term outcomes. Programs combining speech-language therapy, physical therapy, and occupational therapy consistently improve motor milestones, communication skills, and adaptive function. The critical window during early brain development makes early enrollment in intervention programs a priority from the first weeks of life.
Educational outcomes have improved dramatically with inclusive education models. Most children with Down syndrome benefit from placement in general education classrooms with appropriate support. The majority of adults with Down syndrome can achieve semi-independent or supported independent living, hold meaningful employment with support, form lasting friendships, and participate fully in community life. Many drive, read, use technology independently, and live in their own homes or supervised group settings.
Behavioral characteristics often noted include friendliness, sociability, and a strong sense of humor — though behavioral challenges including stubbornness and inattention can be present, and attention-deficit/hyperactivity disorder (ADHD) co-occurs in 6–20% of children with Down syndrome.
Medical Complications
Down syndrome is associated with a range of medical complications that require systematic surveillance throughout the lifespan. The American Academy of Pediatrics publishes health supervision guidelines specifically for Down syndrome, which define age-specific screening schedules.
Thyroid disease is the most common endocrine complication, affecting 15–30% of individuals over a lifetime. Congenital hypothyroidism occurs at higher rates than in the general population and is detected by newborn screening. Acquired hypothyroidism, typically autoimmune (Hashimoto's thyroiditis), accumulates with age. Annual TSH screening is recommended throughout life. Hyperthyroidism is less common but does occur.
Obstructive sleep apnea (OSA) is remarkably prevalent, estimated at 50–80% of individuals with Down syndrome. Contributing factors include hypotonia of the upper airway musculature, macroglossia, midface hypoplasia, and obesity. Untreated OSA worsens cognitive function, behavioral problems, and cardiovascular health. Polysomnography is recommended for all children with Down syndrome by age 4 and whenever symptoms suggest OSA.
Hearing loss affects 50–70% of individuals, predominantly conductive hearing loss secondary to recurrent otitis media and small ear canals, though sensorineural loss also occurs. Prompt treatment of hearing impairment is critical because it directly impacts language development and cognitive outcomes.
Leukemia risk is elevated 10–20-fold above the general population. Two distinct forms are notable: transient myeloproliferative disorder (TMD) occurs in approximately 10% of newborns with Down syndrome and usually resolves spontaneously within 3 months, though it can be fatal and may predict later leukemia. Acute megakaryoblastic leukemia (AMKL) is the most common form of acute myeloid leukemia in young children with Down syndrome, while acute lymphoblastic leukemia (ALL) also occurs at elevated rates. Notably, Down syndrome leukemia is often exquisitely sensitive to chemotherapy, with cure rates exceeding those of leukemia in the general population.
Additional complications include celiac disease (5%), seizure disorders (8–10%), obesity, strabismus and refractive errors (in the majority), cataracts (both congenital and acquired), Hirschsprung's disease (1–3%), and duodenal atresia — a gastrointestinal malformation producing the classic "double bubble" sign on prenatal ultrasound, present in 2–5% of Down syndrome births.
Alzheimer's Disease Connection
The link between Down syndrome and Alzheimer's disease (AD) is one of the most important and well-established connections in human genetics. Because chromosome 21 carries the gene encoding amyloid precursor protein (APP), individuals with Down syndrome — who have three copies of chromosome 21 — produce approximately 50% more APP protein throughout their lives. This excess APP is cleaved into amyloid-beta peptides that accumulate as amyloid plaques, the defining neuropathological hallmark of Alzheimer's disease.
The neuropathological consequences are predictable and devastating in their timing:
- By age 40, virtually all individuals with Down syndrome show Alzheimer's-type neuropathology on autopsy or brain imaging, including both amyloid plaques and neurofibrillary tangles
- Clinical dementia develops in approximately 50% by age 60
- By age 70, approximately 75% of individuals with Down syndrome have clinical Alzheimer's dementia
The clinical presentation of AD in Down syndrome often differs from late-onset AD in the general population. Rather than memory loss as the first symptom, individuals with Down syndrome more commonly present with behavioral changes, personality shifts, loss of previously acquired adaptive skills, and regression in daily living abilities. Seizure onset or worsening is also common in AD-related decline in Down syndrome. Because baseline cognitive assessment is complicated by pre-existing intellectual disability, diagnosis requires comparison to the individual's own prior functioning rather than to population norms.
Research into Alzheimer's disease in Down syndrome has broader implications for the general population. The Down syndrome population provides a unique model for studying the natural history of amyloid accumulation and the pre-clinical phase of AD — potentially decades of biomarker changes before dementia onset.
Current therapeutic research focuses on several targets:
- BACE inhibitors — to reduce APP cleavage and amyloid-beta production
- Anti-amyloid monoclonal antibodies (lecanemab, donanemab) — under investigation in DS-specific trials
- DYRK1A inhibitors — targeting this trisomic kinase that phosphorylates tau and contributes to both cognitive deficits and AD pathology
- The Alzheimer's Biomarker Consortium — Down Syndrome (ABC-DS) and the LuMind IDSC Foundation are leading longitudinal studies to characterize biomarker trajectories and enable clinical trial readiness
Prenatal Screening and Diagnosis
Prenatal identification of Down syndrome has evolved substantially over the past two decades, shifting from primarily second-trimester biochemical screening to highly sensitive first-trimester and cell-free DNA approaches. Screening and diagnostic options should be offered to all pregnant individuals regardless of age, with counseling that clearly distinguishes between screening (probability estimate) and diagnosis (definitive karyotype).
Cell-free fetal DNA (cfDNA) / Non-invasive prenatal testing (NIPT) analyzes fragments of placental DNA circulating in maternal blood from approximately 10 weeks gestation. For trisomy 21, cfDNA achieves:
- Sensitivity exceeding 99% (detection rate)
- False-positive rate below 0.1%
- Positive predictive value that varies with maternal age and population prevalence — approximately 91% at age 25, above 99% at age 40
cfDNA is considered first-line screening in high-risk populations and is increasingly used as population-level screening. A positive cfDNA result requires confirmation by diagnostic karyotyping before any irreversible decision is made.
First-trimester combined screening (11–13 weeks) combines ultrasound measurement of nuchal translucency (NT) with maternal serum PAPP-A (pregnancy-associated plasma protein A) and free beta-hCG. This achieves approximately 85–90% detection with a 5% false-positive rate. Increased NT (≥3.5 mm) is also associated with congenital heart defects and other chromosomal conditions.
Second-trimester quad screen (15–20 weeks) measures maternal serum AFP (alpha-fetoprotein), hCG, unconjugated estriol, and inhibin A. In Down syndrome, the characteristic pattern is low AFP + low estriol + elevated hCG + elevated inhibin A. Detection rate is approximately 80% with a 5% false-positive rate.
Diagnostic confirmation requires fetal karyotyping:
- Chorionic villus sampling (CVS) — 10–13 weeks; provides first-trimester diagnosis; procedure-related pregnancy loss approximately 0.5–1%
- Amniocentesis — 15–20 weeks; considered gold standard for karyotyping; procedure-related loss approximately 0.1–0.3%
Both procedures provide sufficient cells for full karyotype, which confirms the specific cytogenetic type (free trisomy, translocation, or mosaic) — information essential for recurrence risk counseling.
Management and Therapies
There is currently no disease-modifying treatment that addresses the underlying trisomy 21. Management focuses on systematic health surveillance, early intervention, and prompt treatment of associated conditions. The American Academy of Pediatrics health supervision guidelines for Down syndrome provide the standard framework, defining age-specific screening schedules from birth through adulthood.
Newborn period:
- Karyotype confirmation (cytogenetics)
- Echocardiography for all newborns regardless of clinical findings
- Newborn hearing screen
- Thyroid function (TSH) as part of newborn metabolic screen
- Hematologic evaluation (complete blood count) to screen for transient myeloproliferative disorder (TMD)
- Ophthalmologic examination
Early childhood: Enrollment in early intervention programs (speech-language therapy, physical therapy, occupational therapy) as early as possible — ideally within the first months of life. Annual TSH, audiologic screening every 6 months until age 3 then annually, annual ophthalmologic exam. Lateral cervical spine X-rays by age 3–5 to assess for atlantoaxial instability before contact sports or elective surgery requiring neck positioning.
Ongoing across the lifespan: Annual thyroid function tests; sleep study (polysomnography) by age 4 and whenever OSA symptoms arise; regular cardiology follow-up for those with repaired CHD; dental care (malocclusion, periodontal disease common); weight management; annual behavioral/mental health screen. Adults require additional surveillance for Alzheimer's disease — baseline neuropsychological testing at age 30–35 recommended.
Therapies and educational supports: Augmentative and alternative communication (AAC) devices benefit many individuals with limited verbal output. Supported employment programs, transition planning beginning in adolescence, and person-centered care coordination improve adult quality of life. Many individuals with Down syndrome benefit from supported decision-making rather than full guardianship — an approach that respects autonomy while providing necessary support.
Research pipeline: Active clinical trials are investigating DYRK1A inhibitors (to improve cognition by normalizing kinase activity from the trisomic gene), anti-amyloid therapies for the Alzheimer's component, and gene silencing strategies targeting specific chromosome 21 genes. While no disease-modifying therapy is currently approved, the Down syndrome research field has accelerated considerably since the establishment of dedicated NIH and INCLUDE (Investigating Co-occurring conditions across the Lifespan to Understand Down syndromE) research programs.
Key Research Papers
-
Weijerman ME et al. (2008) — Prevalence, neonatal characteristics, and first-year mortality of Down syndrome: a national study. J Pediatr.
Search PubMed -
Bull MJ; AAP Committee on Genetics (2011) — Health supervision for children with Down syndrome. Pediatrics.
Search PubMed -
Roizen NJ, Patterson D (2003) — Down's syndrome. Lancet.
Search PubMed -
Sherman SL et al. (2007) — Epidemiology of Down syndrome: evidence for a new age-related nondisjunction mechanism. Free Radic Biol Med.
Search PubMed -
Freeman SB et al. (1998) — Population-based study of congenital heart defects in Down syndrome. Am J Med Genet.
Search PubMed -
Wiseman FK et al. (2009) — Down syndrome — recent progress and future prospects. Hum Mol Genet.
Search PubMed -
Zigman WB, Lott IT (2007) — Alzheimer's disease in Down syndrome: neurobiology and risk. Ment Retard Dev Disabil Res Rev.
Search PubMed -
Lott IT, Dierssen M (2010) — Cognitive deficits and associated neurological complications in individuals with Down's syndrome. Lancet Neurol.
Search PubMed -
Norton ME et al. (2015) — Cell-free DNA analysis for the detection of fetal trisomy 21 in a general-risk population. N Engl J Med.
Search PubMed -
Savva GM et al. (2010) — An analysis of the relationship between intellectual disabilities and maternal age in England. PLoS One.
Search PubMed -
Bittles AH et al. (2007) — The changing birth prevalence of Down syndrome and the changing risk factors for nondisjunction. Paediatr Perinat Epidemiol.
Search PubMed -
Whooten R et al. (2018) — Endocrine manifestations of Down syndrome. Curr Opin Endocrinol Diabetes Obes.
Search PubMed
Connections
- Genetics
- Inheritance: Dominant, Recessive and Punnett Squares — interactive animation
- Meiosis: Making Eggs and Sperm — interactive animation
- Mitosis: How One Cell Becomes Two — interactive animation
- Turner Syndrome
- Klinefelter Syndrome
- Pediatrics
- Cardiology — Congenital Heart Disease
- Neurology
- Hematology — Leukemia Risk
- Endocrinology — Hypothyroidism