Autoimmune Pancreatitis
- Overview
- Type 1 vs Type 2 AIP — Two Distinct Diseases
- IgG4-Related Disease — The Systemic Connection
- Clinical Presentation — Mimicking Pancreatic Cancer
- Imaging Features — Sausage Pancreas and Duct Changes
- Diagnosis — HISORt Criteria
- Treatment — Steroids and Relapse Prevention
- Complications and Long-Term Course
- Research Papers
- Connections
- Featured Videos
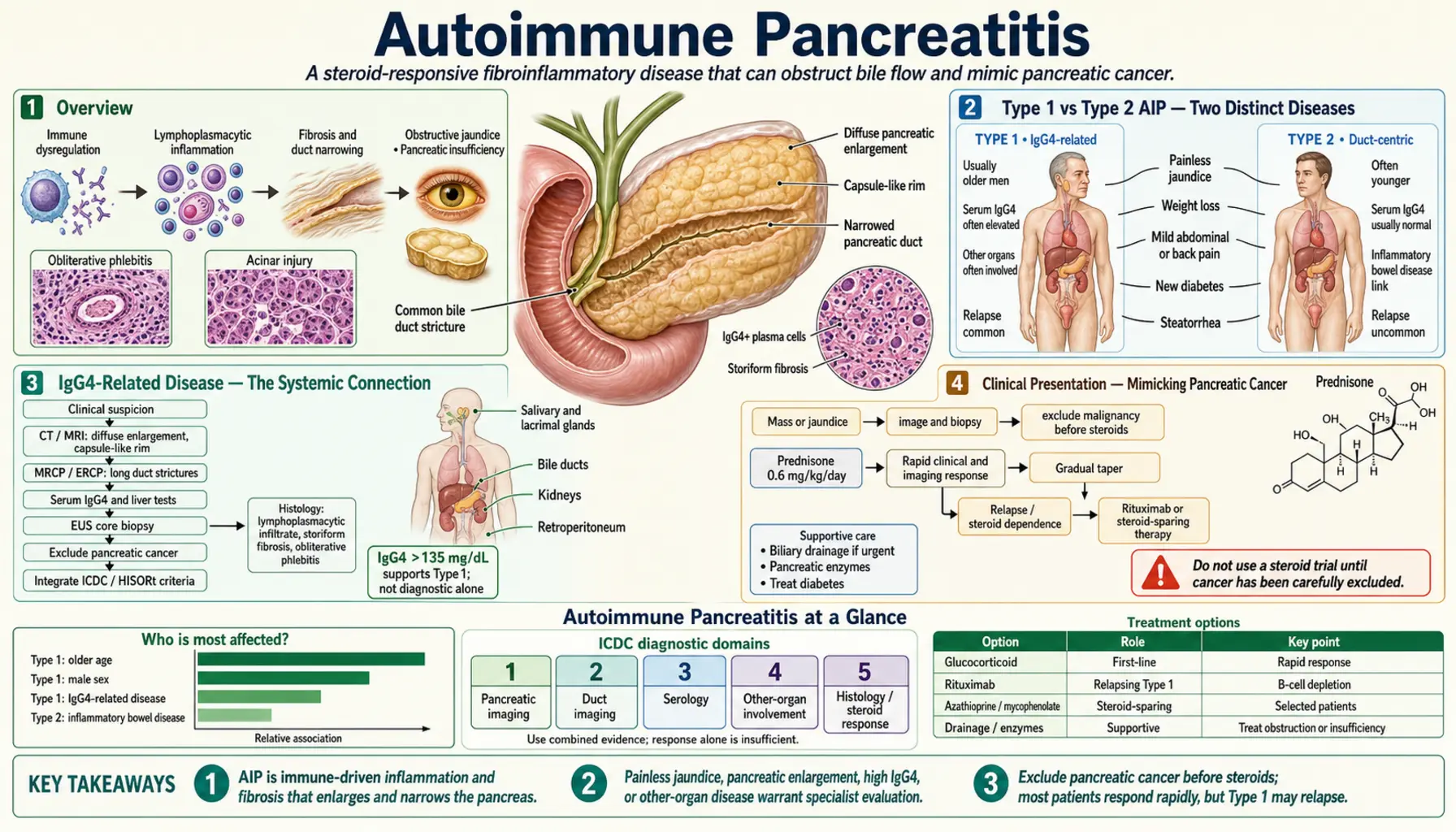
Overview
Autoimmune pancreatitis (AIP) is an immune-mediated fibroinflammatory disease of the pancreas that behaves nothing like the gallstone or alcohol-driven pancreatitis most people know. Rather than causing the sudden, severe abdominal pain that characterizes typical acute pancreatitis, AIP typically causes painless obstructive jaundice — and it does so in a way that so closely resembles pancreatic cancer that it has led to unnecessary major surgery in patients who would have responded completely to a short course of steroids.
The central clinical lesson of AIP is this: before operating on what appears to be a pancreatic head mass with biliary obstruction, consider autoimmune pancreatitis. A trial of corticosteroids — with rapid, dramatic improvement as the expected response — can spare patients a pancreaticoduodenectomy (Whipple procedure) for a benign, steroid-responsive disease.
AIP is not a single disease. Two clinically and pathologically distinct subtypes exist, united by their response to immunosuppression but differing in their underlying mechanism, affected patient population, associated conditions, relapse behavior, and histological fingerprint. Understanding this distinction changes both how you diagnose AIP and how aggressively you manage it long-term.
AIP is rare — estimated prevalence of approximately 2 to 4 per 100,000 population in Japan, where the condition was first systematically described, and somewhat lower in Western countries. Despite its rarity, AIP has garnered enormous clinical interest because of its cancer-mimicking presentation and its complete reversibility with treatment.
Type 1 vs Type 2 AIP — Two Distinct Diseases
The recognition that "autoimmune pancreatitis" encompasses two biologically separate conditions was one of the most important refinements in gastroenterology over the past two decades. The 2011 International Consensus Diagnostic Criteria (ICDC) formalized this distinction.
Type 1 AIP — IgG4-Related Pancreatitis: Type 1 is the more common form, accounting for approximately 60–80% of AIP cases in Western countries and an even higher proportion in Japan and Korea. It is the pancreatic manifestation of IgG4-related disease (IgG4-RD), a systemic fibro-inflammatory condition that can affect virtually any organ. Pathological hallmarks are dense lymphoplasmacytic infiltration with abundant IgG4-positive plasma cells (>10 per high-power field), storiform (swirling, cartwheel-pattern) fibrosis, obliterative phlebitis (inflammatory destruction of veins), and granulocyte epithelial lesion (GEL) absence. These four histological features — lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis, IgG4+ plasma cells — together define LPSP (lymphoplasmacytic sclerosing pancreatitis), the histological name for Type 1 AIP.
Type 1 AIP primarily affects older men — median age at diagnosis 60–70 years, with a male-to-female ratio of approximately 3–4:1. Serum IgG4 is elevated (above the upper limit of normal) in approximately 60–80% of Type 1 patients; levels greater than twice the upper limit of normal are highly specific (>97%) for IgG4-related disease, though sensitivity is only about 56%. Approximately 30–40% of Type 1 patients relapse after stopping steroids, and some require long-term maintenance immunosuppression or rituximab for relapse-prone disease.
Type 2 AIP — Idiopathic Duct-Centric Pancreatitis (IDCP): Type 2 is less common and distinct in every important way from Type 1. Its defining histological feature is the granulocytic epithelial lesion (GEL) — neutrophilic infiltration and destruction of pancreatic duct epithelium. Serum IgG4 levels are normal. The systemic IgG4-RD context is absent. Patients are younger — median age 40–50 years — and the male predominance of Type 1 is absent or reversed. Most strikingly, Type 2 AIP has a strong association with inflammatory bowel disease (IBD): approximately 25–35% of Type 2 patients have Crohn's disease or ulcerative colitis, a rate far exceeding what is seen in Type 1. Type 2 AIP responds well to steroids and, critically, has a very low relapse rate — most patients achieve durable remission after a single course of prednisolone. Long-term maintenance immunosuppression is rarely required.
Why this distinction matters in practice: A young patient with jaundice, new-onset diabetes, and a pancreatic mass who also has active ulcerative colitis should raise immediate suspicion for Type 2 AIP rather than pancreatic cancer. An older man with jaundice, elevated serum IgG4, dry eyes, and enlarged submandibular glands should prompt evaluation for Type 1 AIP and IgG4-RD. Getting the subtype right guides the expected relapse risk and long-term management strategy.
IgG4-Related Disease — The Systemic Connection
IgG4-related disease (IgG4-RD) is the systemic condition of which Type 1 AIP is one manifestation. Recognizing IgG4-RD transformed understanding of AIP — explaining why the "autoimmune pancreatitis" patient often has problems far beyond the pancreas.
What is IgG4-RD? IgG4-RD is a fibro-inflammatory condition characterized by tumefactive (mass-forming) lesions at affected sites, dense lymphoplasmacytic infiltration with abundant IgG4-positive plasma cells, storiform fibrosis, and — in many cases — elevated serum IgG4. It can affect virtually any organ and was recognized as a unified disease entity only in the early 2000s, after Japanese researchers noticed that the same histological pattern kept appearing in patients diagnosed with seemingly unrelated conditions: Riedel's thyroiditis, Küttner's tumor (submandibular gland sclerosis), Mikulicz disease (bilateral lacrimal and salivary gland enlargement), retroperitoneal fibrosis, and interstitial nephritis. All turned out to be the same disease wearing different disguises.
Organs affected in IgG4-RD (relevant to Type 1 AIP):
- Biliary tree — IgG4-related sclerosing cholangitis (IgG4-SC): The biliary complication most relevant to AIP. IgG4-SC causes thickening and stricturing of bile ducts, potentially affecting the intrahepatic and extrahepatic bile ducts, and mimics primary sclerosing cholangitis (PSC) or cholangiocarcinoma. When biliary involvement accompanies pancreatic AIP, the stricturing of the common bile duct causes obstructive jaundice — the most common presenting symptom of Type 1 AIP.
- Salivary and lacrimal glands — Mikulicz disease: Bilateral, painless enlargement of the parotid, submandibular, and lacrimal glands. Presents clinically as dry eyes and dry mouth (sicca symptoms). On clinical grounds nearly indistinguishable from Sjögren's syndrome, but the histology, serology (IgG4 elevated, ANA/anti-Ro/La negative), and steroid response differ.
- Kidneys — IgG4-related tubulointerstitial nephritis: Interstitial inflammatory infiltrate rich in IgG4-positive plasma cells, often causing renal cortical lesions visible on imaging (wedge-shaped low-attenuation areas on CT). Can cause renal insufficiency. Responds to steroids.
- Retroperitoneum — IgG4-related retroperitoneal fibrosis: Dense fibrosis encasing the aorta, inferior vena cava, and ureters. Can cause ureteral obstruction and hydronephrosis.
- Lymph nodes, aorta, thyroid, lung, prostate, meninges: Less common but recognized sites of IgG4-RD involvement.
Serum IgG4 interpretation: IgG4 is the least abundant of the four IgG subclasses (IgG1/2/3/4) in normal individuals. Normal serum IgG4 is typically less than 135 mg/dL (though the upper limit of normal varies by laboratory). In IgG4-RD, levels may be modestly elevated (1–2× ULN) or markedly elevated (>10× ULN). The critical thresholds are: >1× ULN = suspicious; >2× ULN = highly specific for IgG4-RD when combined with appropriate clinical and radiological findings. Important caveat: approximately 5–10% of patients with undoubted IgG4-RD (confirmed histologically) have normal serum IgG4 — a negative serum IgG4 does not exclude the diagnosis. Conversely, IgG4 can be mildly elevated in pancreatic cancer, primary sclerosing cholangitis, eosinophilic granulomatosis, and other conditions — a positive serum IgG4 alone is insufficient to diagnose IgG4-RD.
Pathogenesis of IgG4-RD: Still incompletely understood. Current evidence points to an aberrant T helper 2 (Th2) and T regulatory (Treg) immune response, with overproduction of IL-4, IL-5, IL-10, and IL-13, and excessive TGF-beta driving the characteristic fibrosis. IgG4 itself may not be the pathogenic effector — it is more likely a bystander marker of B cell activity. Plasmablasts (CD19+CD38+CD20-) are the best serum marker for disease activity and rituximab (anti-CD20) efficacy, driving its use in relapsing Type 1 AIP.
Clinical Presentation — Mimicking Pancreatic Cancer
The clinical presentation of AIP is best understood through the framework of what it most dangerously resembles — and what it does not look like.
What AIP looks like:
- Obstructive jaundice (most common presentation, ~70%): Painless, progressive yellowing of the skin and eyes with dark urine and pale stools — the classic triad of biliary obstruction. In the context of a pancreatic head mass on imaging, this presentation is near-impossible to distinguish from pancreatic adenocarcinoma without further workup. The biliary obstruction in AIP results from either inflammatory swelling of the pancreatic head compressing the distal common bile duct, or from IgG4-SC causing stricturing of the biliary tree (Type 1).
- New-onset diabetes mellitus: Occurs in 40–80% of AIP patients at diagnosis. Both exocrine and endocrine pancreatic function are disrupted by the diffuse inflammatory process. AIP-related diabetes may be partially or completely reversible with steroid treatment — improvement in diabetes after steroids is a useful diagnostic clue and a meaningful therapeutic goal. Persistent diabetes post-treatment suggests irreversible beta-cell loss.
- Steatorrhea and exocrine insufficiency: Impaired pancreatic enzyme secretion from diffuse parenchymal inflammation causes fat malabsorption. Patients may report greasy, floating, difficult-to-flush stools. Pancreatic enzyme replacement therapy (PERT) treats the exocrine component.
- Weight loss: From a combination of exocrine insufficiency, reduced caloric intake due to early satiety or anorexia, and the metabolic effects of underlying inflammation. Weight loss contributes to the cancer-mimicking picture.
- Mild abdominal discomfort: Unlike typical acute pancreatitis, severe abdominal pain is not a feature of AIP. Mild epigastric discomfort may be present, but the absence of the severe, boring pain that characterizes acute pancreatitis is diagnostically informative.
- IBD symptoms (Type 2 AIP): Bloody diarrhea, rectal urgency, or known Crohn's disease in a younger patient presenting with AIP features should immediately shift suspicion toward Type 2.
What AIP does NOT look like:
- Severe, acute epigastric pain radiating to the back — the hallmark of acute pancreatitis from gallstones or alcohol. AIP does not cause acute pancreatitis flares.
- Elevated serum lipase or amylase to the degree seen in acute pancreatitis — pancreatic enzyme elevations in AIP are mild if present at all.
- Signs of sepsis or systemic inflammation — AIP is smoldering and fibroinflammatory, not acutely infectious.
The cancer mimic problem in practice: Studies have documented that 2–5% of patients undergoing Whipple resection (pancreaticoduodenectomy) for presumed pancreatic cancer are found to have AIP on pathological review of the resected specimen. This represents an enormous harm — major surgery with significant morbidity and mortality for a disease that would have resolved with prednisone. The practical upshot: in any patient with a pancreatic head mass and biliary obstruction, particularly when classic cancer features (vascular involvement, metastases, CA19-9 extremely high) are absent, AIP should be rigorously considered and a steroid trial may be diagnostically and therapeutically appropriate.
Imaging Features — Sausage Pancreas and Duct Changes
Imaging plays a central role in AIP diagnosis — not to confirm AIP definitively (tissue biopsy does that), but to identify features that support AIP over pancreatic cancer and allow a safer diagnostic path.
CT and MRI of the pancreas:
- "Sausage-shaped" pancreas (diffuse form): The most recognizable imaging finding in AIP. The pancreas loses its normal lobulated, feathery contour and becomes diffusely enlarged, smooth, and cylindrical — resembling a sausage on axial imaging. This diffuse enlargement is characteristic enough that the term "sausage-shaped pancreas" is used in major diagnostic criteria. It occurs in approximately 40–50% of AIP cases (more common in Type 1).
- Peri-pancreatic halo ("rim sign" or "capsule-like rim"): A thin rim of low attenuation (on CT) or T2 hyperintensity (on MRI) surrounding the enlarged pancreatic parenchyma. Represents the inflammatory fibrous capsule encasing the gland. This rim or halo is specific to AIP and is rarely seen with pancreatic cancer — its presence should strongly suggest AIP.
- Focal form — pancreatic head mass: In approximately 30–40% of AIP cases, the inflammation is focal rather than diffuse, creating a discrete mass in the pancreatic head, body, or tail. This focal mass is the direct cause of the cancer-confusion problem. Focal AIP mass tends to lack the vascular encasement (superior mesenteric artery, celiac axis involvement) that characterizes pancreatic ductal adenocarcinoma.
- Upstream pancreatic duct dilation — characteristically absent or mild: One of the most important distinguishing features from pancreatic cancer. In pancreatic ductal adenocarcinoma obstructing the pancreatic duct, the upstream duct dilates markedly (the "double duct sign" — simultaneous dilation of the main pancreatic duct and common bile duct). In AIP, even when a mass is present in the head causing biliary obstruction, the upstream main pancreatic duct is typically NOT markedly dilated — the inflammatory stricture is long and gradual rather than abrupt.
MRCP and ERCP — pancreatic duct findings: The pancreatic duct itself shows characteristic changes on MRCP (magnetic resonance cholangiopancreatography) or ERCP (endoscopic retrograde cholangiopancreatography):
- Long, irregular stricture: AIP characteristically produces a long, irregular narrowing of the main pancreatic duct involving more than one-third of the duct length. The narrowed segment is not smoothly tapered but irregular and beaded.
- Multiple non-contiguous strictures: May be present in diffuse AIP.
- Side branches arising from the strictured segment: The visibility of side branches from within the strictured area is a feature suggesting AIP rather than cancer (cancer destroys the duct architecture).
- Biliary involvement: ERCP or MRCP may show bile duct stricturing from IgG4-SC — intrahepatic and extrahepatic duct involvement — in addition to the pancreatic duct findings.
18F-FDG PET/CT: AIP typically shows marked FDG avidity in the pancreas (and in other IgG4-RD-affected organs). This can be misleading as pancreatic cancer is also FDG-avid. However, PET/CT can identify extrapancreatic IgG4-RD manifestations (salivary gland, kidney, retroperitoneal involvement) that would support AIP diagnosis.
Diagnosis — HISORt Criteria
The HISORt criteria, developed at the Mayo Clinic and published in 2006, provide a structured framework for diagnosing AIP when the constellation of clinical, radiological, serological, and histological findings is evaluated together. HISORt stands for Histology, Imaging, Serology, Other organ involvement, and Response to steroids.
H — Histology: Core needle biopsy of the pancreas (EUS-guided fine-needle biopsy preferred) can show the characteristic histological features of LPSP (Type 1) or IDCP (Type 2). Histology is the most definitive diagnostic criterion but is not always safely obtainable. Whipple resection specimens provide the most tissue but represent overtreatment if done for AIP diagnosis alone.
I — Imaging: CT/MRI showing the "sausage-shaped" pancreas with peri-pancreatic rim, OR ERCP/MRCP showing the long irregular pancreatic duct stricture as described above. Imaging findings must be consistent with AIP, not merely non-specific.
S — Serology: Elevated serum IgG4 (>140 mg/dL = 1× ULN; >280 mg/dL = 2× ULN is highly specific). Normal serum IgG4 does not exclude AIP, particularly Type 2. Anti-lactoferrin antibodies and anti-carbonic anhydrase II antibodies were described in early AIP literature but are not routinely used clinically. For Type 2 AIP, no specific serum marker exists.
O — Other organ involvement: Extrapancreatic IgG4-RD manifestations (sclerosing cholangitis, salivary gland enlargement, renal lesions, retroperitoneal fibrosis) strongly support Type 1 AIP. IBD (Crohn's disease or ulcerative colitis) supports Type 2 AIP. Evidence of other organ involvement can be detected clinically, serologically, or on imaging.
Rt — Response to steroids: The classic diagnostic maneuver when other criteria are insufficient to exclude pancreatic cancer confidently. A trial of prednisolone (0.5–0.6 mg/kg/day, approximately 40 mg/day for 2 weeks) with repeat imaging is offered when AIP is the leading diagnosis but tissue is unavailable. Rapid, dramatic radiological improvement — near-resolution of the pancreatic mass and normalization of the biliary stricture within 2 weeks — essentially confirms AIP. Absence of response or clinical deterioration raises serious concern for malignancy and mandates urgent tissue diagnosis.
2011 International Consensus Diagnostic Criteria (ICDC): The most widely used international system, developed by a consortium including Japanese, Korean, European, and North American gastroenterologists. The ICDC assigns criteria in five categories (P = parenchymal imaging, D = ductal imaging, S = serology, OOI = other organ involvement, H = histology) as Level 1 (highly specific) or Level 2 (less specific) findings for each AIP subtype. Diagnosis of Type 1 or Type 2 AIP requires specific combinations of these criteria, with or without response to steroid therapy as a supplementary criterion. The ICDC provides the most nuanced framework for distinguishing Type 1 from Type 2 AIP.
When to biopsy vs. trial steroids: The decision requires clinical judgment. If malignancy cannot be excluded with reasonable confidence based on HISORt/ICDC criteria, tissue biopsy should be obtained before steroids — because steroids can cause temporary radiological improvement even in lymphoma, which is another important differential diagnosis for a pancreatic mass with elevated IgG4. EUS-guided fine-needle biopsy (FNB) using a core biopsy needle has become the preferred method for obtaining adequate tissue for histological diagnosis without surgical risk.
Treatment — Steroids and Relapse Prevention
The treatment of AIP is one of the most satisfying in gastroenterology — a patient presenting with what looks like incurable pancreatic cancer achieves near-complete resolution within weeks on oral prednisone. Understanding the treatment arc — induction, taper, monitoring for relapse, and rescue therapy — is essential for long-term management.
Initial steroid therapy (induction): The standard regimen is prednisolone 0.5–0.6 mg/kg/day (approximately 30–40 mg/day) for 4 weeks, followed by a gradual taper. The most widely used taper protocol reduces the dose by 5 mg every 1–2 weeks, with a total treatment duration of approximately 3–6 months before attempting complete cessation. In patients with obstructive jaundice requiring biliary drainage (endoscopic biliary stenting), steroids are initiated after biliary decompression — though some guidelines support concurrent treatment if the clinical diagnosis of AIP is confident.
Response monitoring: Repeat imaging (CT or MRI) at 2 weeks of treatment is the standard practice when steroids are used diagnostically. Clinical response — reduction in jaundice, improved diabetes, weight gain — should also be assessed. Serum IgG4 levels fall with treatment in Type 1 AIP (useful for monitoring) but do not normalize completely in all patients and should not guide steroid discontinuation alone.
Relapse:
- Type 1 AIP: High relapse rate — approximately 30–50% relapse after initial steroid course, typically within 3 years. Relapse most commonly involves the biliary tree (IgG4-related sclerosing cholangitis) or pancreas itself. Risk factors for relapse: proximal biliary involvement at diagnosis, persistent elevated serum IgG4 after treatment, diffuse (rather than focal) pancreatic involvement at presentation.
- Type 2 AIP: Low relapse rate — the vast majority of patients achieve durable remission after a single steroid course. IBD activity does not reliably predict AIP relapse.
Maintenance immunosuppression for relapsing Type 1 AIP: Options include low-dose prednisolone maintenance (2.5–7.5 mg/day for 1–3 years), azathioprine (2 mg/kg/day), or mycophenolate mofetil. Japanese guidelines traditionally favored long-term low-dose steroid maintenance; Western practice has moved toward steroid-sparing agents or rituximab for relapse-prone patients.
Rituximab (anti-CD20 B-cell depletion): The most important advance in AIP management for steroid-dependent or frequently relapsing Type 1 patients. Rituximab (1000 mg IV × 2 doses, 2 weeks apart, or 375 mg/m² × 4 weekly doses) achieves remission in approximately 90% of relapsing Type 1 AIP patients. Serum IgG4 levels fall dramatically after rituximab. Retreatment at first signs of serological or clinical relapse (rising IgG4, re-emergence of symptoms) is highly effective. Rituximab has essentially replaced the need for long-term daily steroid maintenance in most relapsing patients at experienced centers.
Exocrine and endocrine management: Pancreatic enzyme replacement therapy (PERT — lipase, amylase, protease) for exocrine insufficiency. Diabetes management; insulin may be required acutely. Post-treatment recovery of endocrine function is variable — roughly 50% of AIP-related diabetes partially or completely resolves with steroid treatment; the remainder requires ongoing antidiabetic therapy. If diabetes persists 12 months after achieving AIP remission, recovery is unlikely.
Biliary interventions: Temporary biliary stenting (endoscopic or percutaneous) for severe obstructive jaundice at presentation, bridging to steroid response. Long-term biliary stenting is rarely required if AIP responds to steroids. In IgG4-SC with dominant bile duct strictures that do not respond to steroids (rare), biliary reconstruction may be necessary.
Complications and Long-Term Course
AIP is often portrayed as a benign, steroid-responsive condition — and in many patients it is. But the long-term course can involve significant complications, and the disease carries real morbidity if not properly managed.
Pancreatic exocrine insufficiency (PEI): Even after successful AIP treatment and resolution of pancreatic inflammation, fibrosis from the inflammatory process can leave residual exocrine insufficiency. Studies report PEI in approximately 50–80% of AIP patients at long-term follow-up, often requiring ongoing PERT. Regular monitoring of nutritional status and fat-soluble vitamin levels is important.
Diabetes mellitus: AIP-associated diabetes may persist indefinitely if extensive beta-cell destruction occurred during the inflammatory episode. Pancreatic exocrine disease-related diabetes (Type 3c diabetes) has distinct pathophysiology from Type 1 or Type 2 diabetes — it involves both insulin deficiency (from beta-cell destruction) and insulin resistance reduction (from glucagon deficiency), making hypoglycemia a particular risk with insulin therapy. Management requires awareness of these distinctive features.
Pancreatic atrophy and duct irregularity: Chronic fibroinflammatory changes can leave the pancreas atrophic and the pancreatic duct irregularly narrowed on imaging, even in well-treated patients. These changes are sequelae of prior inflammation rather than active disease and should not be misinterpreted as treatment failure.
Pancreatic cancer risk: A particularly important and controversial area. Some studies suggest a modestly increased risk of pancreatic malignancy in AIP patients compared to the general population, though the absolute risk remains low. Whether this represents causal association, surveillance bias (AIP patients undergo more imaging), shared risk factors, or misclassification of early cancer as AIP in some cases is debated. Current evidence does not support intensive pancreatic cancer surveillance programs specifically for AIP, but clinical vigilance is appropriate, particularly in patients with recurrent episodes or atypical features.
IgG4-RD progression (Type 1): Untreated or inadequately treated Type 1 AIP can be complicated by progressive IgG4-RD affecting other organs — particularly biliary (IgG4-SC leading to secondary biliary cirrhosis), renal (tubulointerstitial nephritis leading to chronic kidney disease), and retroperitoneal (retroperitoneal fibrosis causing ureteral obstruction). Regular surveillance for extrapancreatic IgG4-RD involvement is a part of Type 1 AIP long-term management.
Quality of life: Studies consistently show impaired quality of life in AIP patients compared to healthy controls, driven by fatigue, ongoing diabetes management, dietary adaptations for exocrine insufficiency, and anxiety about cancer risk and disease recurrence. Patient education and access to multidisciplinary support significantly improve outcomes.
Research Papers
- PMID 16931569 — Chari ST, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4(8):1010–6. doi: 10.1016/j.cgh.2006.05.017. — Search PubMed
- PMID 21849971 — Shimosegawa T, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352–8. doi: 10.1097/MPA.0b013e3182142fd2. — Search PubMed
- PMID 22931785 — Sah RP, Chari ST. Autoimmune pancreatitis: an update on classification, diagnosis, natural history and management. Curr Gastroenterol Rep. 2012;14(2):95–105. doi: 10.1007/s11894-012-0246-2. — Search PubMed
- PMID 23935912 — Kamisawa T, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58(11):1504–7. doi: 10.1136/gut.2008.172908. — Search PubMed
- PMID 25921430 — Hart PA, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut. 2013;62(12):1771–6. doi: 10.1136/gutjnl-2012-303617. — Search PubMed
- PMID 25684593 — Kamisawa T, et al. IgG4-related disease. Lancet. 2015;385(9976):1460–71. doi: 10.1016/S0140-6736(14)60720-0. — Search PubMed
- PMID 26303120 — Buijs J, et al. Pancreatic function after recovery from autoimmune pancreatitis. Dig Liver Dis. 2015;47(12):1056–61. doi: 10.1016/j.dld.2015.07.158. — Search PubMed
- PMID 29439534 — Majumder S, Chari ST. Chronic pancreatitis. Lancet. 2016;387(10031):1957–66. doi: 10.1016/S0140-6736(16)00097-0. — Search PubMed
- PMID 30602458 — Carruthers MN, et al. Rituximab for IgG4-related disease: a prospective open-label trial. Ann Rheum Dis. 2015;74(6):1171–7. doi: 10.1136/annrheumdis-2014-206605. — Search PubMed
- PMID 31270519 — Löhr JM, et al. United European Gastroenterology (UEG) and European Society of Gastroenterology and Endoscopy (ESGE) recommendations for the diagnosis and treatment of autoimmune pancreatitis. United European Gastroenterol J. 2017;5(5):661–72. doi: 10.1177/2050640616671261. — Search PubMed
- PMID 33972082 — Kamisawa T, et al. Japanese Clinical Guidelines for Autoimmune Pancreatitis 2018. Pancreatology. 2020;20(6):1091–100. doi: 10.1016/j.pan.2020.03.010. — Search PubMed
- PMID 36261898 — Bossi P, et al. Autoimmune pancreatitis — current updates. Dig Dis Sci. 2022;67(5):1763–75. doi: 10.1007/s10620-022-07702-2. — Search PubMed
PubMed topic searches:
- Autoimmune pancreatitis diagnosis treatment
- IgG4-related disease pancreas sclerosing cholangitis
- Autoimmune pancreatitis type 1 type 2 LPSP IDCP
- Autoimmune pancreatitis steroid treatment relapse
- Rituximab IgG4 related disease autoimmune pancreatitis
Connections
- Gastroenterology
- Pancreatitis (acute and chronic)
- Primary Sclerosing Cholangitis (biliary differential)
- Primary Biliary Cholangitis (biliary autoimmune)
- Inflammatory Bowel Disease (Type 2 AIP association)
- Diabetes (AIP-related Type 3c diabetes)
- Pancreatic Cancer (critical differential diagnosis)
- Gallbladder Disease
- Colorectal Cancer
- Liver Disease