Porphyria Cutanea Tarda (PCT)
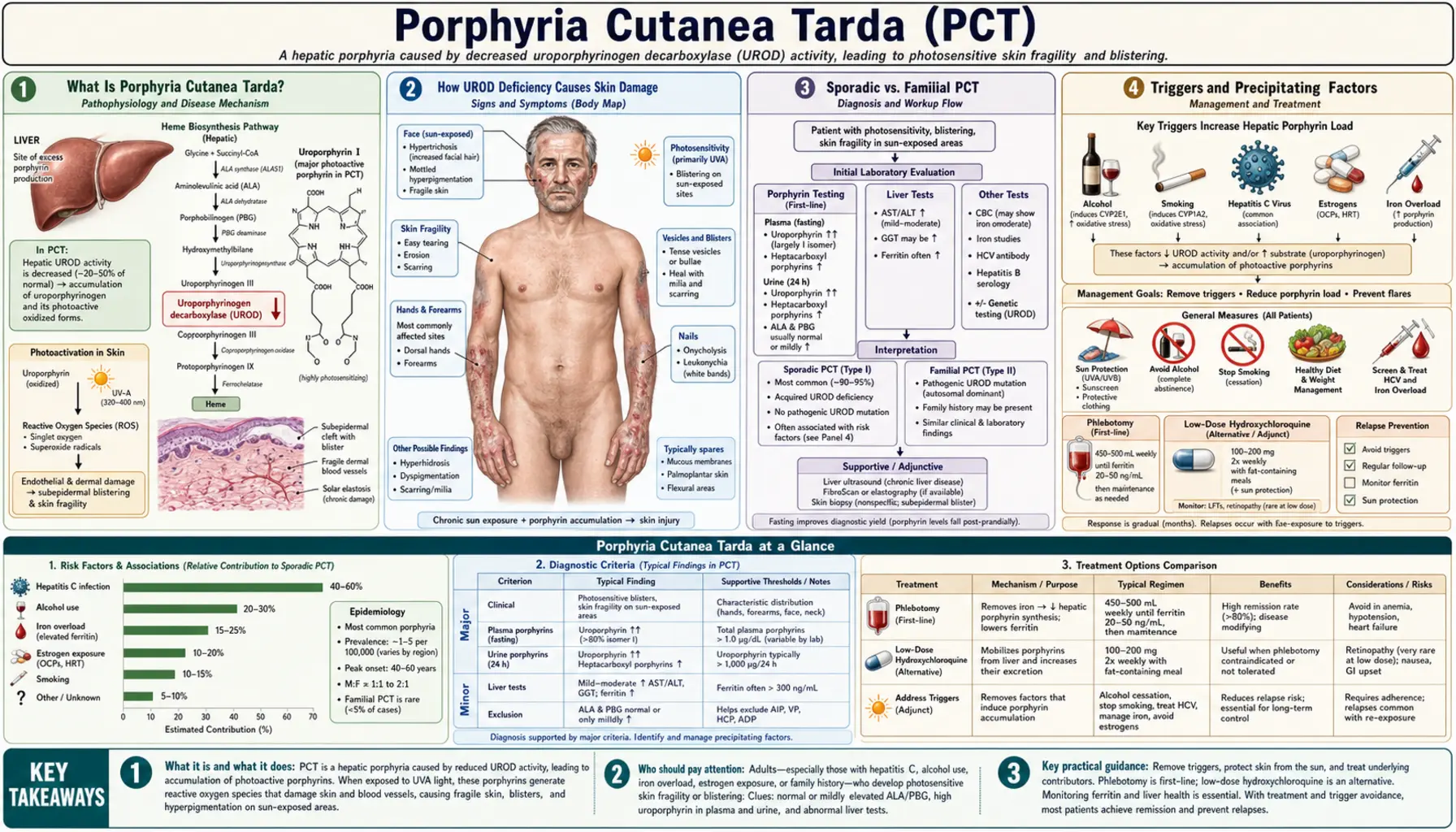
Porphyria Cutanea Tarda is the most common of all the porphyrias — a family of metabolic disorders affecting the heme biosynthesis pathway. In PCT, a deficiency of the enzyme uroporphyrinogen decarboxylase (UROD) causes photoreactive porphyrins to build up in the liver, spill into the bloodstream, and deposit in the skin. When sunlight hits these porphyrins, it triggers a damaging photochemical reaction that produces blistering, scarring, and disfiguring skin changes on sun-exposed areas. The disease is deeply intertwined with treatable triggers — alcohol, hepatitis C, iron overload, and estrogens — meaning that in most patients, identifying and removing the cause can lead to full remission without the need for complex medications.
- What Is Porphyria Cutanea Tarda?
- How UROD Deficiency Causes Skin Damage
- Sporadic vs. Familial PCT
- Triggers and Precipitating Factors
- Skin Findings and Symptoms
- Diagnosing PCT
- Treatment: Removing Triggers and Clearing Porphyrins
- Phlebotomy and Hydroxychloroquine in Detail
- Hepatitis C, HFE Mutations, and PCT
- Prognosis and Long-Term Outlook
- Key Research Papers
- Connections
What Is Porphyria Cutanea Tarda?
Porphyrias are a group of eight distinct disorders, each caused by a deficiency of one of the eight enzymes in the heme biosynthesis pathway. Heme — the iron-containing core of hemoglobin and of cytochrome enzymes throughout the body — is assembled from simpler molecules through this sequential eight-step pathway in the liver and bone marrow. When any one enzyme is deficient, the upstream precursors (porphyrins and their precursors) accumulate, overflow into the blood, and cause disease in specific organs depending on which enzyme is affected and how toxic the accumulating compound is.
PCT affects the fifth enzyme in the pathway: uroporphyrinogen decarboxylase (UROD). When UROD activity falls below approximately 20% of normal in the liver, the enzyme's substrates — uroporphyrins and heptacarboxylporphyrins — back up dramatically and leak into the plasma. These specific porphyrins are photosensitizers: they absorb UV and visible light at approximately 400–410 nanometers (the Soret band) and, when excited by light, transfer that energy to surrounding molecules in a way that generates reactive oxygen species and destroys nearby tissue. In skin, this photochemical reaction severs dermal-epidermal connections, causing the characteristic blistering.
PCT is the most common porphyria worldwide, with an estimated prevalence of approximately 1 in 10,000, though many mild cases go unrecognized. It is an acquired condition in the majority of patients (sporadic PCT) — the enzyme deficiency is not inherited but rather induced by specific environmental and metabolic triggers in genetically predisposed individuals. A minority (about 20%) carry a germline UROD mutation that halves their baseline enzyme activity, making them far more susceptible to these triggers. PCT is not a cancer, not contagious, and not a rapidly fatal disease — but untreated, it causes progressive skin disfigurement and liver damage, and it signals the presence of serious underlying conditions that require their own management.
How UROD Deficiency Causes Skin Damage
The connection between a liver enzyme deficiency and blistering skin rashes is not immediately obvious — it requires understanding the specific photochemical properties of the porphyrins that accumulate and the route by which they reach the skin.
The UROD Enzyme and Its Substrates
In normal heme synthesis, UROD converts uroporphyrinogen III to coproporphyrinogen III through a series of four successive decarboxylation steps, producing a cascade of partially decarboxylated intermediates: heptacarboxylporphyrinogen → hexacarboxylporphyrinogen → pentacarboxylporphyrinogen → coproporphyrinogen. When UROD activity in the liver is suppressed, these intermediates — particularly uroporphyrinogen and heptacarboxylporphyrinogen — accumulate. These compounds are readily auto-oxidized in the presence of iron and oxygen to their porphyrin (oxidized) forms: uroporphyrin I and III, and heptacarboxylporphyrin.
Iron as the Critical Cofactor
Iron is central to PCT pathogenesis and is the reason that iron overload is a consistent finding and phlebotomy is the most effective treatment. Hepatic iron, particularly in the ferric (Fe³⁺) state, appears to generate an oxidized inhibitor of UROD — likely a uroporphomethene or a related oxidized porphyrin compound — that directly and irreversibly inactivates the enzyme. This means iron overload and elevated hepatic iron do not simply co-exist with PCT; they actively drive the enzyme deficiency. Reducing hepatic iron through phlebotomy removes the inhibitory compound, restores UROD activity, and allows porphyrins to normalize.
From Liver to Skin: The Photosensitizer Route
Uroporphyrins and heptacarboxylporphyrins accumulating in the hepatocyte cytoplasm are released into bile and plasma. Plasma porphyrins circulate freely and deposit in sun-exposed skin, particularly in the dermis where small blood vessels carry them close to the surface. When UV or visible light (particularly the 400–410 nm Soret band, which penetrates window glass) strikes these porphyrin-laden skin cells, a Type I and Type II photochemical reaction occurs: singlet oxygen and free radicals are generated, which oxidize proteins, lipids, and nucleic acids in the dermal-epidermal junction. This destroys the anchoring proteins connecting the epidermis to the dermis, producing subepidermal blistering — mechanically similar to, but pathologically distinct from, the autoimmune blistering of bullous pemphigoid.
Liver Damage and Fibrosis
The accumulated porphyrins are not simply cosmetically problematic — they are hepatotoxic. Chronic porphyrin accumulation in the liver drives oxidative stress, inflammation, and hepatic fibrosis. The same triggers (alcohol, hepatitis C) that precipitate PCT are independently hepatotoxic, compounding the liver injury. Long-term uncontrolled PCT substantially increases the risk of hepatocellular carcinoma, and liver surveillance is a component of PCT management in patients with established liver disease.
Sporadic vs. Familial PCT
PCT is classified into two forms based on whether the UROD deficiency is inherited or acquired. This distinction influences counseling, family screening, and the threshold at which disease manifests.
Type I — Sporadic PCT (80% of Cases)
The majority of PCT patients have no inherited UROD mutation. Their germline UROD gene is normal — they have two functional copies — and the enzyme deficiency is entirely acquired, localized to the liver, and caused by the action of environmental and metabolic triggers (alcohol, iron, HCV, estrogens, aromatic hydrocarbons) on the hepatic UROD enzyme. The enzyme inhibitor formed in this process is present only in the liver, not in red blood cell precursors in the bone marrow. When triggers are removed and iron is depleted, UROD activity in these patients fully recovers and porphyrin levels normalize. Sporadic PCT patients have no increased baseline risk of the disease in the absence of triggers, and their children do not inherit an elevated risk (unless the child independently acquires the same triggers and the gene variants that increase susceptibility).
Type II — Familial PCT (20% of Cases)
In familial PCT, one copy of the UROD gene carries a loss-of-function mutation, inherited in an autosomal dominant pattern. This baseline heterozygosity means UROD enzyme activity starts at approximately 50% of normal in all tissues — liver, bone marrow, red blood cells, everywhere. With this head start toward the critical threshold of enzyme insufficiency, the same environmental triggers that might be harmless in a person with two normal UROD copies push the familial PCT patient below the disease threshold more easily. Type II PCT patients have lower thresholds for disease activation, tend to manifest earlier in life, and — importantly — have measurably reduced UROD activity in their red blood cells (a reliable diagnostic marker, unlike sporadic PCT where red blood cell UROD is normal). Their children have a 50% chance of inheriting the UROD mutation, though inheriting the mutation does not guarantee developing PCT (full clinical penetrance still requires exposure to a precipitating trigger). Genetic counseling and family screening are appropriate when Type II is diagnosed.
Type III — Familial Without UROD Mutation
A rare subgroup of familial PCT (Type III) shows familial clustering of PCT without a detectable UROD germline mutation. These families likely share genetic variants that increase susceptibility to trigger-induced UROD inhibition — perhaps HFE hemochromatosis mutations, CYP1A2 polymorphisms, or other modifier genes — without a structural UROD defect. Type III is poorly characterized.
Triggers and Precipitating Factors
PCT is almost never a spontaneous disease. In virtually all patients, the biochemical deficiency is unmasked or worsened by one or more identifiable environmental, metabolic, or infectious triggers. Identifying and removing triggers is the first and most important step in management — and in many patients, trigger removal alone is sufficient to achieve remission.
Alcohol — The Most Common Trigger
Alcohol is the most frequent precipitant of PCT in developed countries. Multiple mechanisms converge: alcohol promotes hepatic iron accumulation by upregulating iron absorption and impairing iron export; alcohol generates hepatic oxidative stress through acetaldehyde and reactive oxygen species; and alcohol suppresses UROD enzyme activity directly. The relationship is dose-dependent — heavier, longer alcohol consumption causes more severe porphyrin accumulation. Abstinence from alcohol is non-negotiable in PCT management. In patients who abstain completely and have no other triggers, some achieve partial or complete remission from abstinence alone, though phlebotomy is usually still necessary to clear the accumulated hepatic iron.
Hepatitis C Virus (HCV) — A Strikingly Strong Association
HCV infection is found in approximately 50% of PCT patients in European and North American series — a prevalence far higher than in the general population. The mechanism is not fully elucidated but likely involves HCV-induced hepatic iron dysregulation (HCV suppresses hepcidin, the master regulator of iron homeostasis, leading to iron loading even without HFE mutations), combined with HCV-driven liver oxidative stress and inflammation. Strikingly, treating HCV with modern direct-acting antivirals (DAAs) — agents like sofosbuvir/velpatasvir, glecaprevir/pibrentasvir — achieves sustained virological response in nearly 95–99% of patients and in many cases leads to spontaneous PCT remission without phlebotomy, as the liver normalizes iron handling and UROD inhibitor production falls. All PCT patients should be screened for HCV; finding and treating it is simultaneously the most important management step and the most modifiable trigger.
Iron Overload and HFE Hemochromatosis Mutations
Elevated hepatic iron and elevated serum ferritin are almost universal in PCT at the time of diagnosis. HFE gene mutations — particularly C282Y homozygosity and C282Y/H63D compound heterozygosity, which cause hereditary hemochromatosis — are found at substantially increased frequency in PCT patients compared to the general population. These mutations amplify iron loading, lowering the threshold at which triggers can inhibit UROD sufficiently to cause disease. All PCT patients should be tested for HFE mutations; those with C282Y homozygosity may require more aggressive phlebotomy and should be counseled about hereditary hemochromatosis in their families.
Estrogens
Combined oral contraceptive pills and hormone replacement therapy containing estrogen are recognized PCT triggers, particularly in women with other predisposing factors. Estrogens increase hepatic iron mobilization and alter porphyrin metabolism. PCT precipitated by OCPs typically resolves when the pill is stopped, though iron depletion (phlebotomy) is still required to complete remission. Women with PCT on estrogen-containing contraception should transition to progestin-only methods or non-hormonal contraception. HRT in menopausal women with PCT should be discontinued or carefully reconsidered.
HIV Infection
HIV co-infection is found at increased frequency in PCT patients, likely through mechanisms similar to HCV: immune dysregulation, chronic hepatic inflammation, and alteration of iron metabolism. The effect of antiretroviral therapy on PCT activity is less clear than that of HCV treatment with DAAs.
Chlorinated Hydrocarbons
The first large epidemic of PCT ever documented was caused by environmental hexachlorobenzene contamination of wheat grain in Turkey between 1955 and 1959, affecting an estimated 4,000 people. This catastrophic exposure demonstrated that exogenous aromatic hydrocarbons can directly inhibit UROD and trigger epidemic PCT. Occupational exposure to certain chlorinated solvents, dioxins, and polychlorinated biphenyls has been implicated in sporadic cases. Modern PCT in developed countries rarely has this cause, but it established the fundamental principle that exogenous chemicals can precipitate PCT.
Smoking
Smoking is an independent risk factor for PCT, possibly through the pro-oxidant and aryl hydrocarbon receptor-activating compounds in tobacco smoke. The effect is additive with alcohol and other triggers.
Skin Findings and Symptoms
PCT is a purely cutaneous porphyria. Unlike the acute hepatic porphyrias (acute intermittent porphyria, variegate porphyria, hereditary coproporphyria), PCT does not cause neurovisceral attacks — no abdominal pain crises, no neurological manifestations, no psychiatric attacks. All of its disease expression is in the skin, on sun-exposed areas, developing insidiously over months to years rather than suddenly. Patients typically have had subtle skin changes for years before recognizing them as a disease.
Blistering on Sun-Exposed Skin
The hallmark of PCT is tense, fluid-filled blisters (bullae) appearing on sun-exposed areas — most prominently the dorsal hands and fingers, forearms, face, ears, and the V-area of the chest when a shirt collar exposes it. The blisters typically develop after minor sun exposure or trivial trauma (pressure, friction) and are not immediately painful — patients often report finding blisters that they did not feel forming. They rupture to leave shallow erosions, which heal slowly over weeks, leaving behind scars and milia (small, firm, white keratin-filled cysts at healed blister sites — a highly characteristic finding).
Skin Fragility
Skin fragility — the tendency to develop erosions from even trivial mechanical injury — is a prominent early feature. Patients notice skin tears from picking up objects, gardening, or simply putting on gloves. The dorsal hands are particularly fragile. This fragility predates visible blistering in some patients and may be the first clue. The mechanism is photodamage to the dermal-epidermal junction proteins (type IV collagen, laminin) making the skin structurally weak even when blisters have not yet visibly formed.
Hyperpigmentation
Diffuse hyperpigmentation on sun-exposed areas — particularly the face and periorbital regions — develops in established PCT. The pigmentation resembles a tan but is irregular and does not fully fade in winter. Periorbital darkening can be striking, giving a characteristic panda-bear appearance. The mechanism involves porphyrin-induced melanocyte stimulation and post-inflammatory hyperpigmentation from chronic low-grade skin injury.
Hypertrichosis — Increased Facial Hair
One of the most distinctive and diagnostically useful features of PCT is hypertrichosis — abnormal, excessive hair growth — particularly over the temples, cheeks, and sideburn areas in women, and generalized facial hypertrichosis in both sexes. This is not virilization (no clitoromegaly, no voice change, no breast atrophy) — it is a porphyrin-induced stimulation of vellus hair follicles to produce longer, darker, terminal-type hairs. In women, who do not normally have coarse facial hair, this finding is cosmetically distressing and is often the presenting complaint that brings them to medical attention. The hypertrichosis reverses with PCT remission, though slowly.
Sclerodermoid Changes
In chronic, inadequately treated PCT, progressive skin thickening and induration develop over sun-exposed areas — a sclerodermoid pattern that mimics localized scleroderma. The skin becomes waxy, thickened, and tight, particularly over the face, forehead, and hands. Calcinosis (calcium deposits palpable under the skin) can develop within sclerodermoid areas. Unlike scleroderma, PCT's skin thickening is driven by porphyrin-induced collagen stimulation from chronic photodamage rather than by autoimmune fibrosis; it affects only sun-exposed areas and regresses (partially) with treatment. Distinguishing PCT from scleroderma requires porphyrin testing — the clinical appearance alone can be confusing.
What PCT Does NOT Cause
PCT does not cause abdominal pain, peripheral neuropathy, psychiatric symptoms, or cardiovascular complications. These features, if present, suggest a different diagnosis — particularly the acute hepatic porphyrias (acute intermittent porphyria, variegate porphyria) which cause neurovisceral attacks and share some biochemical overlap. PCT also does not cause photosensitivity of the immediate burning/stinging type (as in erythropoietic protoporphyria, EPP, which is agonizing and immediate); PCT's skin damage builds gradually over hours to days after cumulative sun exposure, not within minutes.
Diagnosing PCT
The diagnosis of PCT rests on a combination of characteristic clinical features — blistering, fragility, hypertrichosis, hyperpigmentation on sun-exposed areas — and laboratory confirmation of the specific porphyrin pattern. Clinical suspicion should be high whenever a patient presents with these features, particularly in the context of alcohol use, HCV infection, or iron overload.
Urine Porphyrin Testing — The Diagnostic Cornerstone
A 24-hour urine collection or a spot urine sample sent for quantitative porphyrin fractionation is the most important diagnostic test. In PCT, there is dramatic elevation of uroporphyrins (primarily uroporphyrin I) and heptacarboxylporphyrins, with uroporphyrins substantially exceeding coproporphyrins — a ratio that distinguishes PCT from other porphyrias. In active PCT, 24-hour urine uroporphyrin may be 10–50 times the upper limit of normal. The pink or dark brown discoloration of PCT urine under ordinary light, and its orange-red fluorescence under a Wood's lamp (365 nm UV), reflect this massive porphyrinuria and are useful bedside clues — though formal quantification is always required for diagnosis.
Plasma Porphyrin Fluorescence Spectroscopy
Plasma porphyrin measurement with fluorescence scanning is a highly sensitive and specific test for PCT. The emission peak of PCT plasma at 615–620 nanometers, obtained by scanning plasma fluorescence after excitation at 405 nm, is characteristic and distinguishes PCT from variegate porphyria (peak at 625–627 nm) and erythropoietic protoporphyria (peak at 634 nm). Plasma scanning is increasingly available at reference laboratories and is particularly useful when the clinical picture suggests PCT but urine results are equivocal.
Stool Porphyrins
Stool porphyrin analysis shows elevation of isocoproporphyrins (a PCT-specific finding — isocoproporphyrins are generated because UROD's partial block produces an atypical porphyrin side-product specific to this enzyme deficiency). Elevated fecal isocoproporphyrin is pathognomonic for PCT or hepatoerythropoietic porphyria (HEP, the rare homozygous form of PCT). Stool testing is not routinely required when urine and plasma results are clear but is used in difficult diagnostic cases.
Laboratory Workup for Triggers and Organ Involvement
- Iron studies: serum iron, TIBC, transferrin saturation, serum ferritin — ferritin is usually elevated (often 300–1000 ng/mL); elevated transferrin saturation suggests hereditary hemochromatosis
- HFE gene testing: C282Y and H63D mutations; importantly, compound heterozygosity and C282Y homozygosity both substantially increase PCT risk and iron loading severity
- Hepatitis C serology: anti-HCV antibody and HCV RNA (viral load) — essential in every PCT patient; HCV must be found and treated
- HIV serology
- Liver function tests: ALT, AST, GGT (usually elevated in PCT, reflecting liver porphyrin toxicity and co-existing trigger-related liver disease)
- Liver imaging: ultrasound to assess for hepatic steatosis, fibrosis, or cirrhosis; FibroScan or liver MRI in patients with established liver disease
- Red blood cell UROD enzyme activity: normal in sporadic PCT, reduced to approximately 50% in familial PCT — useful for distinguishing Types I and II
- UROD gene sequencing: if familial PCT suspected; identifies the specific mutation for family counseling
Skin Biopsy
Skin biopsy is not required for diagnosis of PCT when porphyrin testing is diagnostic, but it may be performed when the clinical picture is atypical or when another blistering disorder must be excluded. Biopsy shows subepidermal blisters (the blister is below the epidermis), a festooning pattern of dermal papillae protruding into the blister base (characteristic but not specific to PCT), and PAS-positive hyaline material deposited around superficial dermal blood vessel walls (reflecting porphyrin-induced vessel wall damage). Direct immunofluorescence is negative for immunoglobulins (distinguishing PCT from bullous pemphigoid and dermatitis herpetiformis, which show IgG and IgA deposits respectively).
Key Differential Diagnoses
- Bullous pemphigoid: autoimmune subepidermal blistering; tense bullae on normal and urticarial skin; IgG anti-BMZ antibodies on direct immunofluorescence; no porphyrinuria; no hypertrichosis; responds to steroids
- Dermatitis herpetiformis: intensely pruritic papulovesicles on extensor surfaces; IgA deposits on direct immunofluorescence; associated with celiac disease; no porphyrinuria
- Epidermolysis bullosa acquisita: acquired autoimmune EB; anti-collagen VII antibodies; distinguished by immunofluorescence and porphyrin testing
- Pseudoporphyria: drug-induced PCT-like blistering (NSAIDs, tetracyclines, furosemide, nalidixic acid, voriconazole) with normal porphyrin levels; clinical features identical to PCT; drug history is key; stop the offending drug
- Variegate porphyria: shares photocutaneous features with PCT plus causes acute neurovisceral attacks; distinguished by plasma porphyrin fluorescence peak (625–627 nm vs. 615–620 nm in PCT) and stool protoporphyrin elevation
Treatment: Removing Triggers and Clearing Porphyrins
The treatment strategy for PCT follows a logical sequence: first, identify and eliminate all precipitating triggers; second, deplete the accumulated hepatic iron that is perpetuating UROD inhibition; third, if phlebotomy is not tolerated or effective enough, add low-dose hydroxychloroquine to mobilize porphyrins. In the modern era, treating HCV where present has become an additional powerful treatment lever. With appropriate management, the vast majority of PCT patients achieve complete biochemical and clinical remission.
Trigger Elimination — Non-Negotiable First Step
Treatment cannot succeed if the precipitating factors are not removed:
- Alcohol: complete abstinence. Not reduction — abstinence. Continuing alcohol consumption makes phlebotomy far less effective and may prevent remission entirely.
- Estrogens: stop oral contraceptives (transition to progestin-only or non-hormonal contraception); stop HRT. Consider whether the estrogen exposure is truly necessary and what alternatives exist.
- Iron supplements: stop all iron supplementation, multivitamins containing iron, and red meat if consumption is high.
- Hepatotoxic drugs: review the medication list for other potential liver stressors.
Sun Protection — Immediate Symptom Control
While treatments work to clear porphyrins from the body (a process taking months), sun protection is the only immediate measure that prevents new blister formation. Because uroporphyrins absorb at ~400 nm, standard UV-blocking chemical sunscreens (which protect against 280–320 nm UVB and partially against UVA) do not adequately block the phototoxic wavelengths in PCT. Physical sunscreens containing zinc oxide or titanium dioxide, which reflect and scatter the full visible and UV spectrum, are required. Thick physical sunblock should be applied to all sun-exposed areas daily. Broad-brimmed hats, UV-protective long sleeves, and avoidance of midday sun are essential adjuncts. Patients should also be warned that standard window glass does not block the 400 nm Soret band — blistering can occur from sitting near a sunny window for extended periods.
Treat Hepatitis C
All HCV-positive PCT patients should receive antiviral treatment. Modern direct-acting antivirals (DAAs) — sofosbuvir-based regimens (sofosbuvir/velpatasvir, ledipasvir/sofosbuvir, sofosbuvir/velpatasvir/voxilaprevir) or glecaprevir/pibrentasvir — achieve sustained virological response (SVR12, undetectable HCV RNA 12 weeks after treatment) in 95–99% of patients regardless of genotype. Following SVR, many HCV-positive PCT patients experience progressive PCT remission as liver iron metabolism normalizes and hepatic UROD inhibition recedes. In some patients with HCV-PCT and minimal iron overload, HCV treatment alone achieves full PCT remission without phlebotomy.
Phlebotomy and Hydroxychloroquine in Detail
The two active treatments for PCT — phlebotomy and low-dose hydroxychloroquine — work by different mechanisms and have different risk profiles. Phlebotomy is the first-line treatment for most patients; hydroxychloroquine is used when phlebotomy is not feasible or insufficient.
Phlebotomy — First-Line Treatment
Phlebotomy (therapeutic venesection) removes iron from the body by removing iron-containing red blood cells. Each unit (450 mL) of blood removed contains approximately 200–250 mg of iron. By serially depleting the body's iron stores, phlebotomy reduces hepatic iron to the level at which it can no longer generate the oxidized UROD inhibitor — UROD activity recovers, porphyrin production falls, plasma and urine porphyrins normalize, and skin blistering stops.
Protocol: 450 mL phlebotomy every 2–4 weeks. The target endpoint is a serum ferritin of 15–20 ng/mL — a level of relative iron depletion that is safe but below the threshold that perpetuates UROD inhibition. Most patients require 4–8 phlebotomies to reach this target, though heavily iron-loaded patients (C282Y homozygotes, heavy drinkers with alcoholic liver disease) may need 12 or more. Phlebotomy is not painful beyond the needle stick and can be done at blood donation centers or outpatient clinical settings. Iron studies should be checked every 1–2 phlebotomies to track progress.
Response: urine porphyrins begin falling within 1–2 months of starting phlebotomy. Clinical response (cessation of new blistering) typically lags biochemical improvement by 1–3 months. Full skin healing takes 3–6 months after porphyrin normalization. Scars and milia persist after remission; hypertrichosis gradually recedes over 6–12 months.
Contraindications and cautions: phlebotomy is limited by baseline anemia. Hemoglobin should be checked before each session; phlebotomy should be deferred if hemoglobin falls below approximately 11–12 g/dL. Patients with significant cardiac or respiratory disease should have phlebotomies at a slower pace and in monitored settings.
Low-Dose Hydroxychloroquine — Alternative or Adjunct
Hydroxychloroquine (HCQ) — a 4-aminoquinoline antimalarial — works in PCT by a fundamentally different mechanism from phlebotomy. HCQ forms complexes with uroporphyrins in the liver, dramatically increasing their solubility and mobilizing them from hepatic storage into the plasma, from which they are excreted in the urine. The result is a sharp initial spike in urinary porphyrin excretion (which can temporarily worsen photosensitivity and must be distinguished from disease worsening), followed by progressive porphyrin normalization as hepatic porphyrin stores are depleted.
Critical dosing caveat: HCQ must be used at significantly lower doses than standard clinical uses. Standard HCQ doses for lupus or rheumatoid arthritis (200–400 mg daily) cause acute toxic hepatitis in PCT by releasing too much porphyrin too rapidly from the liver — a dangerous and potentially fatal complication. The PCT dose is 100 mg twice weekly or 200 mg twice weekly — far below lupus dosing. At these low doses, HCQ is safe and effective, achieving PCT remission over 6–12 months. When given at the low dose, the risk of HCQ retinal toxicity (which is dose- and duration-dependent) is minimal, but annual ophthalmological screening is reasonable in patients on long-term therapy.
When to use HCQ over phlebotomy: HCQ is preferred when phlebotomy is contraindicated (baseline anemia, poor venous access, severe cardiac disease) or when phlebotomy has been completed to target ferritin but porphyrins remain elevated. It can also be combined with phlebotomy in severe or refractory cases. Some centers use low-dose HCQ as maintenance therapy after remission to prevent relapse if triggers cannot be fully eliminated.
Hepatitis C, HFE Mutations, and PCT
The intersection of PCT with hepatitis C and iron metabolism disorders deserves dedicated discussion because these associations profoundly affect management, prognosis, and counseling.
The HCV–PCT Relationship
The association between HCV and PCT is so strong that HCV testing is mandatory in every newly diagnosed PCT patient. In North America and Europe, approximately 40–60% of PCT patients are HCV-positive. The mechanisms by which HCV promotes PCT are multifactorial: HCV directly suppresses hepcidin (the liver-derived hormone that governs systemic iron homeostasis) by inhibiting hepcidin gene transcription, leading to iron accumulation even in patients without HFE mutations; HCV induces chronic hepatic inflammation and oxidative stress, promoting UROD inhibitor formation; and HCV may directly impair porphyrin metabolism. The hepcidin suppression pathway means that HCV-PCT patients often have moderately elevated ferritin and elevated transferrin saturation despite lacking the HFE mutations traditionally associated with iron overload — HCV itself is loading them with iron.
The therapeutic implication is powerful: treating and curing HCV with DAAs removes the major driver of hepcidin suppression. As hepcidin levels normalize post-SVR, the liver begins appropriately restricting iron absorption, hepatic iron falls, UROD inhibitor formation drops, and PCT remits — sometimes completely, without any phlebotomy. Studies of PCT patients treated with DAAs before the widespread adoption of phlebotomy show PCT remission rates of 70–90% following SVR alone. In practice, most specialists treat HCV first, then reassess whether phlebotomy is still needed 6–12 months after SVR.
HFE Hemochromatosis and PCT
HFE mutations — particularly C282Y, which in homozygous form causes hereditary hemochromatosis — are found in 10–20% of sporadic PCT patients, substantially exceeding the general population frequency (C282Y homozygosity occurs in approximately 0.3–0.5% of Northern Europeans). HFE mutations amplify iron loading: C282Y homozygotes absorb iron far more aggressively from the gut because of chronically suppressed hepcidin, leading to severe hepatic iron overload. When this iron load is combined with alcohol, HCV, or estrogen exposure, PCT threshold is crossed far more readily. C282Y homozygotes with PCT typically require many more phlebotomies to deplete iron (often 15–30 units over 12–24 months) and should be evaluated and managed according to hereditary hemochromatosis guidelines, including screening of first-degree relatives for HFE mutations.
Hepatocellular Carcinoma Risk
Long-term PCT patients with chronic HCV infection, significant hepatic fibrosis or cirrhosis, or hemochromatosis have substantially elevated risk of hepatocellular carcinoma (HCC). Liver surveillance with ultrasound every 6 months, with or without AFP (alpha-fetoprotein) measurement, is recommended for PCT patients with cirrhosis or advanced fibrosis — the same surveillance protocol used for any chronic liver disease at HCC risk. This is not a consequence of PCT itself but of the underlying liver diseases (HCV, alcohol, hemochromatosis) that drive PCT.
Prognosis and Long-Term Outlook
PCT has an excellent prognosis for skin disease when properly diagnosed and treated. Most patients achieve complete clinical and biochemical remission within 6–18 months of starting appropriate management. The skin blistering stops, hypertrichosis recedes, hyperpigmentation fades over years, and sclerodermoid thickening partially resolves. For most patients, PCT does not shorten life expectancy directly — the skin disease is manageable, reversible, and does not progress to systemic involvement.
Relapse
PCT can relapse if triggers are reintroduced after remission. Patients who resume alcohol, restart estrogen therapy, or develop new HCV infection (or experience failure of HCV treatment) are at risk of biochemical and clinical relapse. Annual monitoring of urine porphyrins and iron studies allows early detection of relapse before clinical blistering recurs. Some patients benefit from continued sun protection and trigger avoidance monitoring indefinitely.
The Prognostic Driver — Liver Disease
The factor that most influences long-term survival in PCT patients is not the porphyria itself but the underlying liver diseases — HCV cirrhosis, alcoholic cirrhosis, and hemochromatosis-related liver fibrosis — that triggered PCT. Patients who achieve HCV cure and stop alcohol have dramatically better liver outcomes than those who do not. The relationship between PCT management and liver disease management is thus synergistic: the steps that eliminate PCT triggers (curing HCV, stopping alcohol, depleting iron) are simultaneously the most effective hepatoprotective interventions available.
Quality of Life in Remission
After remission, most PCT patients can live normal lives with appropriate sun protection as an ongoing precaution. Permanent scarring from past blistering, residual milia, and sometimes residual skin fragility on the hands remain. Patients who achieved PCT remission before developing sclerodermoid changes or significant scarring have the best cosmetic outcomes. The psychological burden of PCT — particularly the hypertrichosis in women, the visible hand scarring, and the lifestyle restrictions imposed by strict sun protection — should not be underestimated; dermatology follow-up and patient support resources are part of comprehensive PCT care.
Key Research Papers
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology. 1998;27(6):1661–1669 — Search PubMed
- Phillips JD, Bergonia HA, Reilly CA, et al. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci USA. 2007;104(12):5079–5084. PMID: 17360334
- Singal AK, Kormos-Hallberg C, Lee C, et al. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2012;10(12):1402–1409. PMID: 22985607
- Jalil S, Grady JJ, Lee C, et al. Associations among behavior-related susceptibility factors in porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2010;8(3):297–302 — Search PubMed
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880–893 — Search PubMed
- Méndez M, Poblete-Gutiérrez P, García-Bravo M, et al. Molecular heterogeneity of familial porphyria cutanea tarda in Spain: characterization of 10 novel mutations in the UROD gene. Br J Dermatol. 2007;157(3):501–507 — Search PubMed
- Andant C, Puy H, Bogard C, et al. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol. 2000;32(6):933–939. PMID: 10898315
- Gisbert JP, García-Buey L, Pajares JM, et al. Prevalence of hepatitis C virus infection in porphyria cutanea tarda: systematic review and meta-analysis. J Hepatol. 2003;39(4):620–627 — Search PubMed
- Egger NG, Goeger DE, Payne DA, et al. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig Dis Sci. 2002;47(2):419–426. PMID: 11855560
- Boffa MJ, Ead RD, Reed P, et al. A double-blind, placebo-controlled, crossover trial of oral vitamin E in porphyria cutanea tarda. Clin Exp Dermatol. 1996;21(3):200–204 — Search PubMed
- Tannapfel A, Stolzel U, Kösters R, et al. Genetic and proliferative markers in porphyria cutanea tarda-associated hepatocellular carcinoma. Virchows Arch. 2001;439(1):36–42 — Search PubMed
- Bonkovsky HL, Barnard GF. Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology. Semin Liver Dis. 1998;18(1):57–65 — Search PubMed
Connections
- Dermatology
- Epidermolysis Bullosa — another blistering skin disorder, but caused by inherited structural protein defects rather than porphyrin accumulation; classified by anatomical level of skin split rather than biochemistry
- Bullous Pemphigoid — autoimmune blistering disease of the elderly; shares tense blisters and subepidermal split with PCT but is driven by IgG autoantibodies against BP180/BP230, not porphyrins; direct immunofluorescence distinguishes the two
- Dermatitis Herpetiformis — gluten-triggered IgA-mediated blistering skin disorder; pruritic vesicles on elbows/knees; distinguished by IgA deposits on DIF and association with celiac disease
- Lichen Sclerosus — chronic inflammatory skin disease causing skin thickening and sclerosis; shares sclerodermoid appearance with advanced PCT but does not blister and has no porphyrin biochemistry
- Morphea — localized scleroderma with skin induration mimicking the sclerodermoid changes of chronic PCT; differentiated by histology and normal porphyrin levels
- Iron — iron overload is the central metabolic driver of PCT; depletion of excess hepatic iron through phlebotomy is the primary treatment mechanism; understanding iron homeostasis is essential to understanding PCT
- Lab Tests — porphyrin fractionation (urine and plasma) and iron studies are the cornerstone diagnostic tests for PCT; fluorescence spectroscopy distinguishes PCT from variegate porphyria