Epidermolysis Bullosa
Epidermolysis bullosa (EB) is a group of rare inherited disorders characterized by extreme fragility of the skin and mucous membranes. Caused by mutations in genes encoding structural proteins that anchor the epidermis to underlying tissue, even minor friction or trauma can cause painful blistering, open wounds, and in severe forms, life-threatening complications. EB spans a wide severity spectrum — from localized hand and foot blistering triggered by exercise, to widespread skin loss beginning at birth that is incompatible with survival into adulthood without intensive medical management.
- Pathophysiology and Structural Proteins
- Classification: EB Simplex, Junctional, Dystrophic, Kindler
- Epidermolysis Bullosa Simplex (EBS)
- Junctional Epidermolysis Bullosa (JEB)
- Dystrophic Epidermolysis Bullosa (DEB)
- Kindler Syndrome
- Squamous Cell Carcinoma: The Leading Cause of Death in Severe DEB
- Diagnosis: Biopsy, Immunofluorescence, and Genetic Testing
- Wound Care and Supportive Management
- Gene Therapy: Beremagene Geperpavec (B-VEC)
- Key Research Papers
- Connections
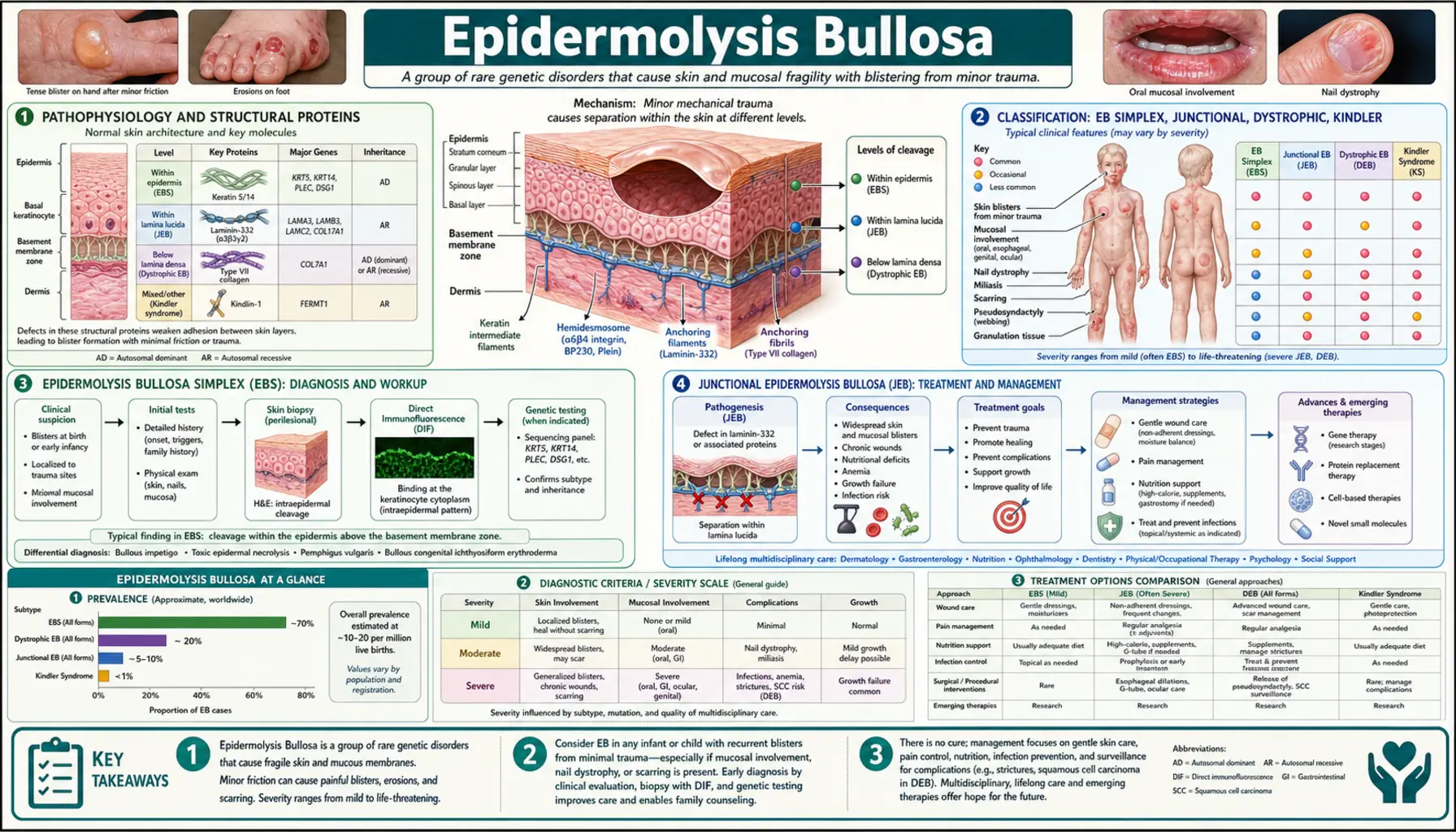
Pathophysiology and Structural Proteins
The skin is held together by a hierarchy of structural proteins that anchor the outer epidermis to the underlying dermis. In healthy skin, keratinocytes in the basal layer are attached to the basement membrane zone (BMZ) through hemidesmosomes, which connect intracellular keratin filaments to the extracellular matrix via transmembrane proteins. Anchoring fibrils made of type VII collagen then secure the basement membrane to the papillary dermis below.
In EB, mutations disrupt one or more of these proteins at a specific anatomical level. The result is a plane of cleavage — a structural weak point within or just beneath the epidermis where mechanical shear forces cause the skin layers to separate, forming a blister filled with tissue fluid. The deeper the cleavage plane, the more severe the consequences: intraepidermal blisters (EB simplex) tend to heal without significant scarring, whereas sub-lamina densa blisters (dystrophic EB) trigger fibroblast activation and lead to progressive scarring, contractures, and organ involvement.
The principal mutated genes and their protein products define each EB subtype:
- KRT5, KRT14 — keratin 5 and keratin 14, cytoskeletal intermediate filaments in basal keratinocytes; mutations cause EB Simplex
- LAMA3, LAMB3, LAMC2 — laminin-332 subunits (alpha-3, beta-3, gamma-2); structural component of the lamina lucida; mutations cause Junctional EB
- COL17A1 — collagen XVII (also called BP180), transmembrane hemidesmosomal collagen; mutations cause Junctional EB; also the autoantigen in Bullous Pemphigoid
- COL7A1 — type VII collagen, the major component of anchoring fibrils beneath the BMZ; mutations cause Dystrophic EB
- FERMT1 — kindlin-1, a focal adhesion protein linking integrins to the actin cytoskeleton; mutations cause Kindler syndrome
Classification: EB Simplex, Junctional, Dystrophic, Kindler
The international classification of EB (2020 consensus) organizes subtypes primarily by ultrastructural level of skin cleavage, determined by transmission electron microscopy (TEM) or immunofluorescence antigen mapping, and confirmed by genetic testing. There are four major types:
- Epidermolysis Bullosa Simplex (EBS) — intraepidermal cleavage (within the basal keratinocyte layer); caused primarily by keratin 5/14 mutations; most common EB type; usually autosomal dominant; blisters heal without significant scarring
- Junctional EB (JEB) — cleavage within the lamina lucida of the basement membrane zone; caused by mutations in laminin-332 or collagen XVII; ranges from lethal (Herlitz subtype) to moderate (non-Herlitz); often autosomal recessive
- Dystrophic EB (DEB) — cleavage below the lamina densa, in the sub-lamina densa zone where anchoring fibrils reside; caused by COL7A1 mutations; scarring is the hallmark; dominant DEB is usually mild, recessive DEB (RDEB) can be severe and life-limiting
- Kindler Syndrome — mixed cleavage planes at multiple levels; caused by FERMT1 mutations; features skin fragility, photosensitivity, progressive poikiloderma, and mucosal involvement
Prevalence of all EB types combined is approximately 19–50 per million live births in population studies. No ethnic or sex predilection distinguishes most forms; Herlitz JEB is uniformly severe regardless of background. Most severe subtypes (RDEB generalized severe, Herlitz JEB) carry a drastically shortened life expectancy without advanced interventions.
Epidermolysis Bullosa Simplex (EBS)
EBS is the most common form of EB, accounting for approximately 70–75% of all cases. The defining molecular event is a missense mutation in KRT5 (keratin 5) or KRT14 (keratin 14), the two intermediate filament proteins that form the keratin network scaffolding inside basal keratinocytes. Mutant keratins assemble into unstable filaments that collapse under mechanical stress, causing the cell itself to rupture — cleavage is therefore within the basal cell, not between the epidermis and dermis.
Most EBS cases follow an autosomal dominant inheritance pattern; only one mutant allele is sufficient to disrupt the keratin network because heterodimers containing one abnormal chain are also structurally compromised. De novo mutations account for a significant proportion. Autosomal recessive EBS (due to null KRT14 mutations) is rarer but often more severe.
Clinical subtypes of EBS range widely in severity:
- EBS Localized (formerly Weber-Cockayne) — the mildest and most common subtype; blistering confined to hands and feet, triggered by friction and warm weather; onset in infancy or early childhood; rarely affects quality of life severely; healing without scarring
- EBS Generalized Intermediate (formerly Koebner) — more widespread blistering on trunk and extremities; present at birth; significant morbidity but not life-limiting
- EBS Generalized Severe (formerly Dowling-Meara) — most severe EBS subtype; widespread herpetiform (grouped) blistering at birth; neonatal period can be life-threatening due to fluid loss and sepsis; improves with age
- EBS with muscular dystrophy — rare; COL15A1 or PLEC mutations; blistering plus progressive late-onset muscular dystrophy
Because blisters in EBS form above the basement membrane, healing occurs without permanent dermal scarring. Milia (small keratin cysts) may form, but the long-term complications of fibrosis seen in dystrophic EB are absent. Management is primarily wound care, blister drainage (to reduce tension), and protective footwear.
Junctional Epidermolysis Bullosa (JEB)
JEB results from mutations in genes encoding proteins of the lamina lucida — the zone between the basal keratinocyte plasma membrane and the lamina densa of the basement membrane. The principal mutated genes are LAMA3, LAMB3, and LAMC2 (encoding the alpha-3, beta-3, and gamma-2 subunits of laminin-332) and COL17A1 (encoding collagen XVII / BP180). Inheritance is autosomal recessive in virtually all cases; two loss-of-function alleles are required.
Two major clinical categories exist:
- Herlitz JEB (JEB severe) — caused predominantly by LAMB3 premature termination codon mutations; the most severe and historically lethal EB subtype; extensive blistering of skin and mucous membranes from birth; characteristic involvement of perioral and perinasal skin with exuberant granulation tissue (hypergranulation tissue around nostrils is pathognomonic); laryngeal/tracheal involvement → airway compromise; urogenital and esophageal strictures; failure to thrive; most affected infants historically died within the first 2 years of life from sepsis, airway obstruction, or malnutrition. With modern intensive multidisciplinary care, some survive longer, but quality of life and prognosis remain extremely poor.
- JEB non-Herlitz (JEB intermediate) — milder mutations that allow residual protein expression; generalized or localized blistering; enamel defects of teeth (hypoplastic enamel is a characteristic clinical clue for JEB); nail dystrophy or nail loss; mucosal involvement less severe than Herlitz; survival into adulthood possible; scarring absent (lamina lucida level) but recurrent erosions cause significant morbidity
Dental enamel defects are clinically useful diagnostic clues for JEB: enamel is derived from the same basement membrane proteins, so laminin-332 mutations simultaneously affect skin and tooth development. Pitted, hypoplastic enamel in a child with blistering should prompt JEB workup.
Dystrophic Epidermolysis Bullosa (DEB)
DEB is caused exclusively by mutations in COL7A1, the gene encoding type VII collagen — the primary structural component of anchoring fibrils that secure the dermis beneath the basement membrane. Because cleavage occurs below the lamina densa in the sub-lamina densa zone, healing involves dermal fibroblast activation and collagen deposition, resulting in scarring as a cardinal feature of all DEB forms.
Two inheritance patterns define the two major groups:
- Dominant DEB (DDEB) — heterozygous glycine substitution mutations in the collagen triple helix domain; dominant negative effect on collagen VII assembly; typically presents as localized or generalized mild disease; characteristic features include albopapuloid lesions (ivory-white papules on trunk), milia, nail dystrophy; life expectancy normal; scarring mild to moderate
- Recessive DEB (RDEB) — homozygous or compound heterozygous loss-of-function mutations; markedly reduced or absent collagen VII; spans from localized RDEB (relatively mild, hands/feet/joints) to RDEB generalized severe (formerly Hallopeau-Siemens) — the most severe and devastating form of all inherited EB
RDEB generalized severe deserves particular attention for the severity of its systemic consequences:
- Pseudosyndactyly — repeated blistering and healing on fingers and toes leads to progressive fusion of digits into "mitten deformities"; hand function lost over years; surgical release provides temporary relief but fusion recurs
- Esophageal strictures — repeated mucosal blistering and scarring narrow the esophagus; dysphagia, aspiration, malnutrition; esophageal dilation under general anesthesia (high-risk in fragile patients) is the main intervention; percutaneous gastrostomy tube placement for nutritional support often required
- Corneal involvement — corneal erosions, scarring, visual impairment
- Anemia — chronic, multifactorial (blood loss from wounds, iron deficiency, inflammation); nearly universal in RDEB generalized severe
- Malnutrition — oral pain limits intake; esophageal strictures worsen it; protein and micronutrient depletion impairs wound healing further
The phrase "butterfly children" refers to the fragility of affected skin — as delicate as a butterfly's wings — though many in the EB community prefer other framing. Patients with RDEB generalized severe require daily full-body wound care requiring hours per day, often performed by caregivers at home.
Kindler Syndrome
Kindler syndrome (also called Kindler-Weary syndrome) is caused by loss-of-function mutations in FERMT1, encoding kindlin-1 — a focal adhesion protein that regulates integrin activation and cytoskeletal-extracellular matrix linkage. Unlike the other EB types where cleavage occurs at a single consistent plane, Kindler syndrome shows cleavage at multiple levels simultaneously (intraepidermal, junctional, and sub-lamina densa), reflecting kindlin-1's role in maintaining structural integrity across multiple skin compartments.
Clinical features that distinguish Kindler syndrome from other EB types include:
- Skin fragility and blistering — present from birth, but often improves with age (atypical among EB types)
- Photosensitivity — actinic damage disproportionate to sun exposure; sunburn-like reactions even with minimal UV exposure
- Progressive poikiloderma — reticulate hyperpigmentation and hypopigmentation with skin atrophy; develops over decades; most prominent on dorsal hands and forearms
- Mucosal involvement — colitis (can mimic inflammatory bowel disease), urethral strictures, esophageal involvement; mucosal disease often drives morbidity in adulthood
- Increased SCC risk — similar to RDEB, though less prominent; driven by chronic wound and scar microenvironment
Inheritance is autosomal recessive. Kindler syndrome is rare, with several hundred reported cases worldwide. The improving natural history of skin fragility with age differentiates it clinically from RDEB, which worsens progressively.
Squamous Cell Carcinoma: The Leading Cause of Death in Severe DEB
In patients with RDEB generalized severe, squamous cell carcinoma (SCC) developing within chronic wounds is the leading cause of premature death. This is not an incidental association — the chronic wound microenvironment of RDEB creates conditions uniquely permissive for malignant transformation:
- Absent or severely reduced collagen VII eliminates normal tumor suppressor signaling from the extracellular matrix
- Chronic wound-associated inflammation generates reactive oxygen species and growth factors (TGF-β, IL-6, VEGF) that promote keratinocyte proliferation and angiogenesis
- Repetitive cycles of destruction and repair select for keratinocytes with acquired mutations in TP53, CDKN2A, and other tumor suppressors
- Impaired immune surveillance in chronically wounded tissue reduces clearance of dysplastic cells
The SCC that develops in RDEB is clinically aggressive and biologically distinct from UV-induced SCC in the general population. Key characteristics:
- Onset typically in the second or third decade of life in RDEB generalized severe
- Often multifocal — several tumors arising simultaneously or in rapid succession
- Rapid local invasion and early metastasis to regional lymph nodes, then distant sites
- Poor response to standard therapies due to the challenging wound environment and patient's systemic fragility
- Cumulative risk reaches 67–90% by age 35 in population-based RDEB studies; median survival after SCC diagnosis is approximately 5 years
Surveillance strategies include systematic dermoscopy of all chronic wounds, low-threshold biopsy of any wound that fails to heal or changes morphology, and regular lymph node assessment. Emerging systemic therapies (immunotherapy with checkpoint inhibitors, targeted agents) are being evaluated in clinical trials specifically for RDEB-associated SCC.
Diagnosis: Biopsy, Immunofluorescence, and Genetic Testing
Accurate diagnosis of EB subtype is essential because prognosis and management differ dramatically between types. Three diagnostic modalities work in concert:
Skin Biopsy with Transmission Electron Microscopy (TEM)
A fresh blister (induced by gentle rubbing of perilesional skin) is biopsied and processed for TEM. The ultrastructural location of the cleavage plane identifies the EB type: within keratinocytes (EBS), within the lamina lucida (JEB), or below the lamina densa (DEB). TEM also allows direct visualization of structural abnormalities (e.g., rudimentary or absent anchoring fibrils in DEB). TEM requires specialized laboratory expertise and is performed at reference centers.
Immunofluorescence Antigen Mapping (IFAM)
Frozen sections of a perilesional biopsy are stained with antibodies against specific basement membrane proteins (collagen IV, collagen VII, laminin-332, collagen XVII/BP180, integrin α6β4). The expression pattern and intensity of each marker identifies the cleavage plane and suggests which protein is deficient. For example:
- Absent or markedly reduced collagen VII staining → DEB (COL7A1 mutation)
- Absent laminin-332 staining → Herlitz JEB (LAMB3/LAMA3/LAMC2 mutation)
- Cleavage below collagen IV (which marks the lamina densa) → sub-lamina densa = DEB
IFAM is faster and more widely available than TEM and has largely replaced TEM as the first-line histological test in many centers.
Genetic Testing
Targeted panel sequencing (all known EB genes) or whole exome sequencing provides the definitive molecular diagnosis. Genetic confirmation enables accurate subtype classification, precise genetic counseling, carrier testing in family members, and eligibility for gene therapy trials. In prenatal diagnosis (for families with a known mutation), chorionic villus sampling (CVS) or amniocentesis allows EB diagnosis before birth.
A practical diagnostic approach: if blistering presents at birth in a neonate, urgent TEM/IFAM biopsy is performed within the first days of life. Clinical features (presence of granulation tissue, dental enamel defects, nail involvement, family history) narrow the differential while awaiting laboratory results.
Wound Care and Supportive Management
There is no cure for inherited EB (with the partial exception of the gene therapy discussed below). Supportive care remains the cornerstone of management across all subtypes and requires a multidisciplinary team: dermatologists, wound care nurses, nutritionists, gastroenterologists, ophthalmologists, orthopedic surgeons, physical and occupational therapists, and palliative care specialists.
Wound Dressings
Standard adhesive bandages and tapes cause tearing in EB skin and are contraindicated. Non-adherent dressings are mandatory:
- Mepitel (silicone-coated polyamide mesh) — primary wound contact layer; does not adhere to wound bed; can be left in place for multiple days
- Mepilex (soft silicone foam) — absorptive secondary layer
- Softsilk / Urgotul — alternatives for primary layer
- Tubular stockinette and cohesive bandage wraps (not adhesive) — outer layers for limbs
Daily or near-daily dressing changes, often taking 1–4 hours for RDEB generalized severe patients, are typically performed by family caregivers at home after training by wound care nurses.
Blister Management
Intact blisters are drained with a sterile needle to reduce pressure and prevent extension (leaving the blister roof in place as a biological dressing). Allowing blisters to expand untreated leads to larger wounds and greater fluid/protein loss.
Nutritional Support
Caloric and protein requirements in severe EB are substantially elevated due to chronic wound losses. Oral nutrition is compromised by oral blistering and esophageal strictures. Supplemental nasogastric or percutaneous gastrostomy tube (PEG) feeding is often required in severe JEB and RDEB. Micronutrient deficiencies (iron, zinc, selenium, vitamin D) are common and require supplementation.
Pain Management
Wound pain in EB is severe and continuous. Multimodal analgesia (topical lidocaine gel for wound care, scheduled analgesics, gabapentinoids for neuropathic components) is essential. Procedural sedation for major wound care or dressing changes is sometimes required in young children.
Gene Therapy: Beremagene Geperpavec (B-VEC)
In 2023, the U.S. FDA approved beremagene geperpavec (trade name Vyjuvek, previously called B-VEC) — the first topical gene therapy approved for any condition and the first approved disease-modifying treatment for EB. This approval represented a landmark milestone for the EB community after decades of research and advocacy.
Mechanism
B-VEC is a replication-incompetent herpes simplex virus type 2 (HSV-2) vector engineered to carry a functional copy of COL7A1 — the gene encoding type VII collagen. The HSV-2 backbone was chosen because herpes simplex virus naturally infects keratinocytes, enabling efficient gene delivery to the target cell type. When applied topically to wounds, the vector enters keratinocytes in the wound bed and delivers the COL7A1 gene, enabling these cells to produce functional type VII collagen and restore anchoring fibrils at the basement membrane zone.
Indication and Clinical Trial Results
B-VEC is approved for topical treatment of DEB (both dominant and recessive) in patients aged 6 months and older caused by COL7A1 mutations. The pivotal Phase 3 randomized controlled trial (GEMINI trial) used a within-patient design: each patient had matched wounds treated with either B-VEC or placebo gel (vehicle). The primary endpoint of complete wound closure at 6 months was achieved in 67% of B-VEC-treated wounds versus 22% of vehicle-treated wounds. New collagen VII deposition at the basement membrane zone was confirmed by immunofluorescence in treated wounds. Adverse effects were generally mild (application site pain, pruritus).
Limitations
B-VEC is a topical therapy applied directly to wounds; it does not address internal mucosal disease, esophageal strictures, pseudosyndactyly, or the systemic wound burden of RDEB generalized severe. The COL7A1 gene delivered is episomal (does not integrate into the genome), so gene expression does not persist indefinitely — repeated applications are required. The therapy addresses the molecular defect in DEB specifically; it has no direct application in JEB or EBS. Cost and access remain significant barriers.
Other Emerging Therapies
- Ex vivo gene-corrected skin grafts — patient keratinocytes harvested, virally transduced with COL7A1 ex vivo, grown into sheets, and grafted onto wounds; limited by scale (can cover only a fraction of total body surface)
- Allogeneic bone marrow / hematopoietic stem cell transplantation (HSCT) — donor-derived cells engraft and some circulating cells contribute to wound healing; modest clinical improvements in RDEB reported; significant transplant-related morbidity and mortality
- mRNA therapy — COL7A1 mRNA encapsulated in lipid nanoparticles for transient collagen VII expression in wounds; Phase 2 trials ongoing
- CRISPR gene editing — preclinical studies correcting COL7A1 mutations in patient-derived iPSCs
Key Research Papers
- Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–1126. PMID: 24690439
- Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–627. PMID: 32017015
- Marinkovich MP, Tang JY. Gene therapy for epidermolysis bullosa. J Invest Dermatol. 2019;139(6):1221–1226 — Search PubMed
- Christiano AM, Greenspan DS, Lee S, Uitto J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J Biol Chem. 1994;269(32):20256–20262. PMID: 8051117
- Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28(1):107–114. PMID: 19945622
- Mellerio JE, Robertson SJ, Bernardis C, et al. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: best clinical practice guidelines. Br J Dermatol. 2016;174(1):56–67 — Search PubMed
- Prodinger C, Reichelt J, Bauer JW, Laimer M. Epidermolysis bullosa: advances in research and treatment. Exp Dermatol. 2019;28(10):1176–1189. PMID: 31136023
- Palisson F, Larralde M. Epidermolysis bullosa in Latin America. Int J Dermatol. 2008;47(4):426–428 — Search PubMed
- Rashidghamat E, McGrath JA. Novel and emerging therapies in the treatment of recessive dystrophic epidermolysis bullosa. Intractable Rare Dis Res. 2017;6(1):6–20. PMID: 28357196
- Nyström A, Bruckner-Tuderman L. Matrix molecules and skin biology. Semin Cell Dev Biol. 2018;89:136–146 — Search PubMed
- Gurevich I, Agarwal P, Zhang P, et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: a Phase 1 and 2 trial. Nat Med. 2022;28(4):780–788 — Search PubMed
- Eichstadt S, Barber C, Larouche J, et al. From clinical phenotype to genotype in epidermolysis bullosa simplex with muscular dystrophy: plectin mutations revisited. Br J Dermatol. 2019;180(6):1478–1486 — Search PubMed
Connections
- Dermatology
- Pemphigus Vulgaris — autoimmune intraepidermal blistering disease; COMPARE: acquired autoimmune vs. inherited structural defect; both cause skin fragility
- Bullous Pemphigoid — autoimmune subepidermal blistering in the elderly; COL17A1 (BP180) is the BP autoantigen and also mutated in JEB
- Psoriasis — chronic inflammatory skin disorder; contrasts with EB (structural vs. immune mechanism) but both involve keratinocyte dysregulation
- Vitiligo — autoimmune pigment loss; distinct mechanism but frequently discussed alongside other rare chronic skin diseases in patient communities
- Pyoderma Gangrenosum — rare ulcerative skin condition; shares chronic wound management challenges with severe EB
- Gastroenterology — esophageal strictures and colitis are major complications of severe JEB and Kindler syndrome
- Oncology — squamous cell carcinoma in chronic RDEB wounds: leading cause of death in severe recessive dystrophic EB