Scleritis
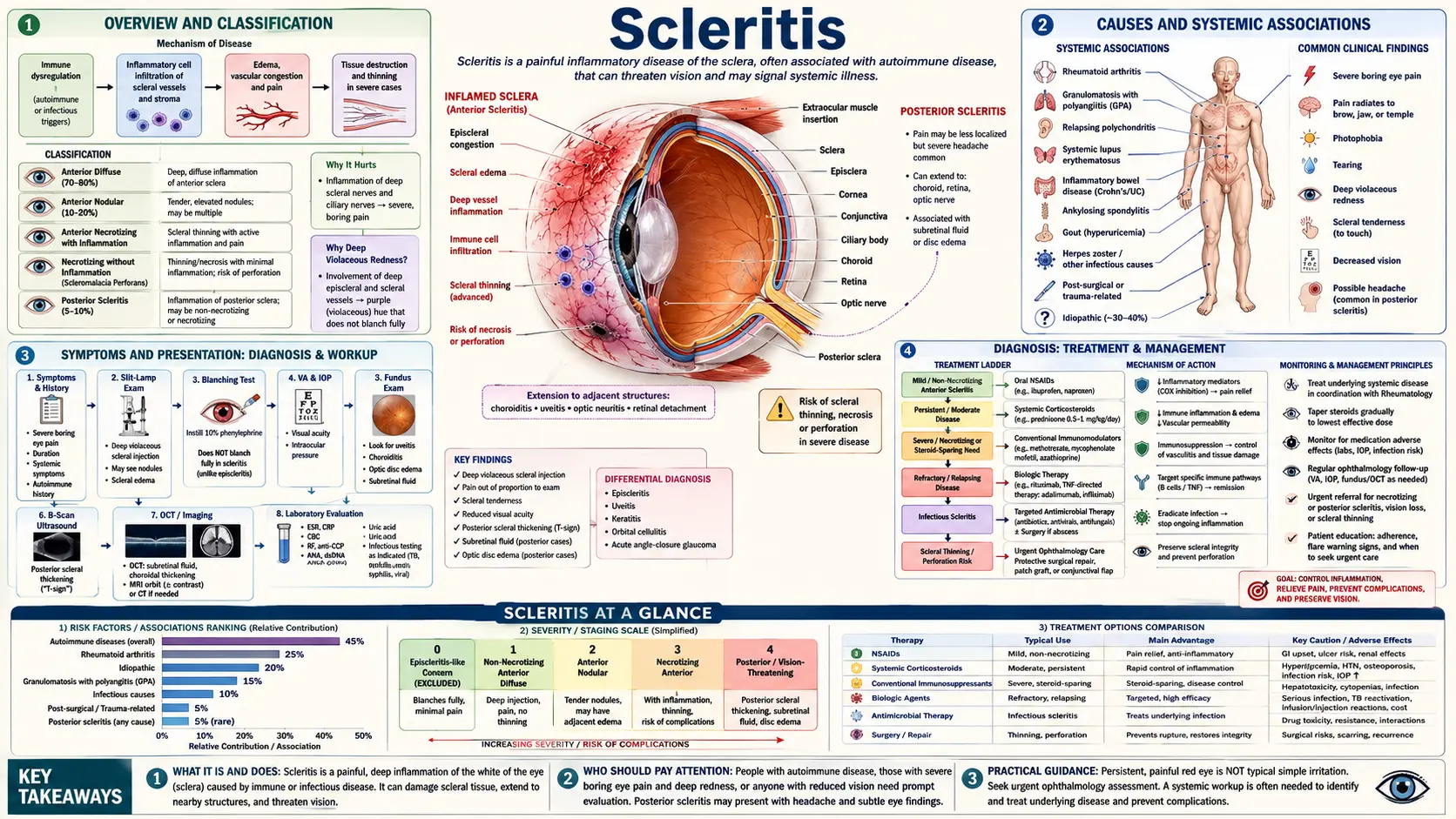
Scleritis is inflammation of the sclera — the tough, white outer coat that forms the structural shell of the eyeball. Unlike common red-eye conditions such as conjunctivitis or a burst blood vessel, scleritis is not a surface irritation. It is a deep inflammatory process that can penetrate all layers of the scleral wall, threaten vision, and in many cases signal a serious systemic autoimmune or rheumatic disease that requires urgent medical evaluation. The hallmark is severe, deep, boring eye pain that often wakes patients from sleep, radiates to the forehead and jaw, and is not relieved by simple pain medications — pain of a character that sets scleritis apart from almost every other eye condition.
Scleritis is uncommon but not rare, affecting roughly 4 to 6 people per 100,000 annually. It tends to appear in middle age and affects women more often than men. Up to half of all cases are linked to an underlying systemic disease — most commonly rheumatoid arthritis, granulomatosis with polyangiitis (formerly Wegener's), or other autoimmune vasculitides. Finding and treating that underlying disease is often as important as treating the eye itself. Without proper management, the most severe subtypes of scleritis can cause permanent scleral thinning, corneal melting, glaucoma, or loss of vision.
When to seek urgent care: Any episode of severe, deep eye pain — especially pain that is constant, radiates into the face, or accompanies a dark-red or violaceous discoloration of the eye — should be evaluated by an ophthalmologist promptly, ideally the same day. Do not assume it is a simple red eye. If your vision is changing, if you have an established autoimmune condition, or if the pain is severe enough to disturb your sleep, go to an urgent eye-care facility or emergency room. Early diagnosis and treatment are the most reliable way to protect your vision.
- Overview and Classification
- Causes and Systemic Associations
- Symptoms and Presentation
- Diagnosis
- Treatment — Anti-Inflammatory
- Treatment — Immunosuppressive and Biologics
- Episcleritis — Diagnosis and Management
- Complications
- Prognosis and Monitoring
- Key Research Papers
- Connections
Overview and Classification
The sclera is a dense collagen-rich coat that gives the eye its shape and protects the delicate internal structures — the lens, vitreous, retina, and optic nerve. It makes up roughly 80% of the outer surface of the globe. When the sclera becomes inflamed, that protective shell is under attack, and because the sclera is not easily replaced or regenerated, progressive inflammation can permanently weaken, thin, or destroy it.
Scleritis is classified primarily by location — anterior or posterior:
- Anterior scleritis involves the sclera in front of the equator of the eye, where it is visible to an examiner under a slit lamp. It accounts for approximately 98% of all scleritis cases and has four recognized subtypes.
- Posterior scleritis involves the sclera behind the equator of the eye, where it cannot be directly visualized on external exam. It is diagnosed by imaging — B-scan ultrasound or MRI of the orbit. It accounts for only about 2% of cases but is frequently misdiagnosed as a tumor, posterior uveitis, or orbital disease.
Anterior Scleritis Subtypes
Diffuse anterior scleritis is the most common subtype, accounting for roughly 40% of anterior cases. There is widespread hyperemia (redness) affecting a broad segment or the entire anterior sclera. The involved vessels have a characteristic deep red to violaceous (purple-red) hue. Despite its alarming appearance, diffuse anterior scleritis is the mildest subtype and is the most likely to respond to oral anti-inflammatory treatment.
Nodular anterior scleritis presents with one or more tender, immobile, inflamed nodules on the surface of the sclera. A critical diagnostic point: a scleritic nodule cannot be moved with a cotton-tipped applicator because it is attached to and arising from the scleral tissue itself. This distinguishes it from episcleritic nodules, which are superficial and can be gently shifted. Nodular scleritis tends to be more persistent and recurrent than the diffuse form but carries a generally good visual prognosis with treatment.
Necrotizing anterior scleritis with inflammation is the most severe and vision-threatening subtype. It is characterized by progressive scleral thinning, necrosis (tissue death), and the appearance of an avascular (bloodless) gray-white patch amid surrounding intense inflammation. The pain is extreme. It is strongly associated with long-standing rheumatoid arthritis, particularly in patients with known systemic vasculitis, and represents an ocular emergency. Without aggressive systemic immunosuppression, it can progress to globe perforation.
Necrotizing scleritis without inflammation (scleromalacia perforans) is a paradoxically painless condition. There is progressive, silent scleral thinning and necrosis with minimal surrounding inflammatory signs. It occurs almost exclusively in patients with severe, long-standing seronegative or seropositive rheumatoid arthritis. Because it is painless, it may go unnoticed until the sclera is severely thinned and the underlying dark uveal tissue becomes visible through the attenuated sclera. Spontaneous globe perforation, while rare, can occur.
Episcleritis: The Important Mimic
Episcleritis — inflammation of the episclera, a thin vascular tissue lying between the conjunctiva and the sclera — is frequently confused with scleritis. The distinction matters enormously, because episcleritis is generally a benign, self-limiting condition that often requires no more than artificial tears and time, while scleritis may require years of systemic immunosuppression.
Key distinguishing features: Episcleritis causes mild discomfort or a gritty aching sensation, not the severe boring pain of scleritis. The redness in episcleritis is typically sectoral (in one area) and the vessels are bright red and can be blanched by instilling phenylephrine 2.5% eye drops — because episcleritic vessels are superficial and responsive to vasoconstrictors. In scleritis, the deep scleral vessels do not blanch with phenylephrine. Episcleritis rarely causes scleral edema, rarely affects vision, and resolves spontaneously in 2 to 3 weeks in the majority of cases. The majority of episcleritis cases have no underlying systemic cause.
Causes and Systemic Associations
The most important thing to understand about scleritis is that in many patients, it is a manifestation of systemic disease rather than a standalone eye problem. Identifying the underlying cause shapes treatment, prognosis, and long-term surveillance.
Idiopathic (No Identified Systemic Cause)
Despite thorough evaluation, no systemic cause is identified in approximately 40 to 50% of scleritis patients. These are classified as idiopathic and treated based on severity and subtype. Even when initially idiopathic, patients should be re-evaluated periodically, as systemic disease can emerge months to years after an initial scleritis diagnosis.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is the single most common systemic disease associated with scleritis, accounting for 30 to 40% of cases linked to a systemic condition. RA-associated scleritis preferentially develops in patients with longstanding, seropositive disease (positive RF and/or anti-CCP antibodies), particularly those with known extra-articular vasculitis. The necrotizing subtypes — both with and without inflammation — are disproportionately associated with RA. Scleritis in an RA patient can be a marker of overall disease activity and increased systemic vasculitis risk; it should prompt reassessment of the patient's rheumatologic management.
Granulomatosis with Polyangiitis (GPA / Wegener's)
GPA is the most common systemic vasculitis to cause scleritis, typically producing the necrotizing subtype. Orbital involvement (proptosis, restricted motility, pain) can coexist. An ANCA (antineutrophil cytoplasmic antibody) titer — specifically c-ANCA / anti-PR3 — is the key serologic marker. GPA-associated scleritis often requires cyclophosphamide or rituximab, not just steroids.
Other Autoimmune Diseases
- Systemic lupus erythematosus (SLE) — associated with anterior and posterior scleritis; ANA, anti-dsDNA, complement levels are the key labs.
- Relapsing polychondritis — bilateral scleritis; can cause corneal melting and hearing loss; characteristic recurrent cartilage inflammation (ears, nose, trachea).
- Inflammatory bowel disease — both Crohn's disease and ulcerative colitis are associated; scleritis activity may parallel bowel flares.
- Reactive arthritis (formerly Reiter's syndrome) — classically the triad of urethritis, arthritis, and conjunctivitis, but scleritis can occur.
- Polyarteritis nodosa — medium-vessel vasculitis; necrotizing scleritis is a recognized manifestation.
- Sarcoidosis — granulomatous inflammation can involve the sclera; chest X-ray and ACE level are screening tools.
- Ankylosing spondylitis — HLA-B27-associated; more commonly causes anterior uveitis but scleritis is reported.
Infectious Causes
Infections are a less common but critical category to identify because they require specific antimicrobial treatment — and treating infectious scleritis with immunosuppression alone, without covering the organism, can be catastrophic.
- Herpes zoster ophthalmicus (HZV) — reactivation of the varicella-zoster virus in the ophthalmic division of the trigeminal nerve; sectoral scleritis is a recognized complication; requires systemic antiviral therapy (acyclovir, valacyclovir).
- Pseudomonas aeruginosa — a feared cause of infectious scleritis in contact-lens wearers and post-surgical patients; rapidly destructive; requires aggressive topical and systemic antipseudomonal therapy.
- Surgically induced necrotizing scleritis (SINS) — an immune-mediated, not truly infectious, reaction occurring months to years after pterygium excision (especially with mitomycin C) or cataract surgery; frequently misdiagnosed as infection; requires systemic immunosuppression.
- Tuberculosis — rare; consider in endemic regions or immunocompromised hosts.
- Syphilis — always exclude with serology (VDRL/RPR + confirmatory FTA-ABS) in every scleritis workup, regardless of clinical suspicion.
- Lyme disease — reported in endemic areas; serology (ELISA + Western blot) for Borrelia burgdorferi.
- Aspergillus and other fungi — rare; almost exclusively in immunocompromised hosts; poor prognosis without antifungal therapy.
Recommended Systemic Workup
All patients with confirmed scleritis — regardless of subtype — should undergo a minimum systemic evaluation. Recommended laboratory panel: complete blood count (CBC), comprehensive metabolic panel (CMP), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), antinuclear antibody (ANA), antineutrophil cytoplasmic antibody (ANCA with c-ANCA/p-ANCA specificity and anti-PR3/MPO), rheumatoid factor (RF), anti-cyclic citrullinated peptide antibody (anti-CCP), VDRL or RPR followed by FTA-ABS (syphilis serology in all cases), uric acid, and ACE level. Chest X-ray is standard. HLA-B27 testing may be added if spondyloarthropathy is suspected. Referral to rheumatology should be arranged for any patient with a positive systemic finding.
Symptoms and Presentation
The Defining Symptom: Deep Boring Pain
The most important and pathognomonic feature of scleritis is the character of the pain. Patients consistently describe it as a severe, deep, constant, boring ache — often comparing it to pressure behind the eye or a sensation that the eye is being pressed from the inside out. The pain radiates along the branches of the trigeminal nerve to the brow, temple, and jaw. It is typically worst at night and will commonly awaken a patient from sleep. It is not relieved by topical anesthetic eye drops, which distinguishes scleral pain from corneal or surface pain. Simple over-the-counter pain relievers provide incomplete relief at best. This severity and quality of pain is unusual for an eye condition, and it should prompt immediate ophthalmologic evaluation.
Eye Appearance
The eye appears red, but the color is distinctive. In scleritis, the redness has a characteristic deep violaceous (bluish-red or purple-red) hue, reflecting congestion in the deeper scleral vessels rather than the superficial conjunctival capillaries. Scleral edema (swelling of the scleral tissue itself) may be present and can be confirmed by slit-lamp examination showing thickening of the scleral stroma. In nodular scleritis, an immobile, tender, reddish nodule is visible on the scleral surface. In necrotizing scleritis, an avascular grayish-white patch — devoid of visible blood vessels — appears within the inflamed area, signaling scleral necrosis.
Associated Ocular Symptoms
- Photophobia (light sensitivity) — often prominent due to reflex ciliary spasm and inflammation.
- Tearing and watering — secondary to pain and inflammation.
- Visual blurring — can arise from associated corneal involvement (sclerosing keratitis), secondary glaucoma, cataract, or cystoid macular edema from posterior segment involvement.
- Restricted eye movement — particularly in posterior scleritis due to involvement of the posterior Tenon's capsule and extraocular muscle sheaths.
Posterior Scleritis — A Diagnostic Trap
Posterior scleritis has a different and potentially misleading presentation because the inflamed sclera is not visible on external examination. Patients may present with pain, proptosis (forward protrusion of the eye), restricted eye movement, choroidal folds (rippling of the retina seen on fundus examination), and exudative retinal detachment — fluid that lifts the retina away from the underlying pigment epithelium without a visible tear. This constellation can mimic an orbital tumor, thyroid eye disease, or uveal melanoma, and the diagnosis is frequently delayed. B-scan ultrasound is essential: it shows the pathognomonic "T-sign," which is fluid accumulation in the posterior Tenon's capsule forming the horizontal bar of the letter T when the optic nerve (vertical) is the stem.
Episcleritis Presentation Contrast
In episcleritis, symptoms are far milder. Patients notice a sectoral or diffuse redness, sometimes with a mild aching or gritty discomfort, but not the severe pain of scleritis. The condition comes on quickly, often within hours, and resolves spontaneously in 2 to 3 weeks without specific treatment in most cases. Vision is almost always normal. There is no scleral edema. Episcleral vessels, when examined at the slit lamp, are bright red (not violaceous), and they blanch when phenylephrine 2.5% drops are instilled.
Diagnosis
Clinical Evaluation at the Slit Lamp
The slit-lamp examination is the cornerstone of scleritis diagnosis. The examiner assesses the depth and character of vessel injection, the presence and mobility of scleral nodules, scleral edema, and the presence of avascular patches (necrosis). A key maneuver: the examiner attempts to gently move the injected vessels with a cotton-tipped applicator. In episcleritis, the superficial vessels can be shifted slightly. In scleritis, the deep scleral vessels are immobile. The examination is performed in both natural light and with a blue cobalt filter to enhance visualization of the deep vascular pattern.
Phenylephrine 2.5% Test
One drop of phenylephrine 2.5% is instilled into the eye. Wait 10 to 15 minutes. If the redness largely blanches, the vessels are superficial (episcleral) and the diagnosis favors episcleritis. If the deep redness persists with only partial surface blanching, the diagnosis is scleritis. This test is quick, inexpensive, and highly useful in the office setting for distinguishing the two conditions, though the distinction should be confirmed by slit-lamp examination.
B-Scan Ultrasonography
B-scan ultrasound is the essential investigation for suspected posterior scleritis. The "T-sign" — fluid accumulation in Tenon's capsule posterior to the globe, visualized alongside the optic nerve shadow — is pathognomonic for posterior scleritis. B-scan also demonstrates scleral-choroidal thickening (greater than 2 mm is considered significant) and can detect associated exudative retinal detachment.
MRI of the Orbit
MRI with gadolinium contrast is the best imaging modality for posterior scleritis when B-scan is equivocal or when orbital mass needs to be excluded. MRI shows scleral thickening and gadolinium enhancement of the scleral-choroidal complex, along with periocular fat stranding. It also provides the best anatomical delineation for planning if surgical biopsy is ever considered.
Optical Coherence Tomography (OCT)
OCT of the posterior pole can reveal choroidal folds, subretinal fluid, and choroidal thickening in posterior scleritis. Enhanced depth imaging OCT (EDI-OCT) is particularly useful for measuring choroidal thickness as a marker of inflammation and treatment response.
Fluorescein Angiography
In severe or necrotizing cases, fluorescein angiography may show choroidal ischemia (areas of non-perfusion in the choroidal vasculature), which correlates with the severity of scleral inflammation and the risk of visual loss.
Differential Diagnosis
Conditions that must be distinguished from scleritis include:
- Episcleritis — milder pain, blanches with phenylephrine, self-limiting.
- Anterior uveitis — pain and photophobia but with anterior chamber flare and cells; perilimbal (ciliary) injection; responds to topical steroids.
- Orbital cellulitis — fever, leukocytosis, proptosis, restricted motility; CT shows periorbital fat stranding and sometimes abscess.
- Conjunctivitis — follicles or papillae, mucopurulent discharge, no significant pain, normal visual acuity.
- Corneal ulcer — pain relieved by topical anesthetic; fluorescein staining reveals epithelial defect.
- Uveal melanoma or metastasis — can mimic posterior scleritis on imaging; biopsy may be needed; a key distinction is that scleritis responds rapidly to systemic steroids while tumor does not.
Treatment — Anti-Inflammatory
Treatment of scleritis follows a stepwise approach based on subtype severity, response to therapy, and the presence or absence of systemic disease. The overarching principle is to control scleral inflammation rapidly enough to prevent structural damage to the eye while managing the underlying systemic condition in parallel.
Oral NSAIDs — First Line for Mild to Moderate Non-Necrotizing Scleritis
Oral nonsteroidal anti-inflammatory drugs (NSAIDs) are the standard initial treatment for diffuse and nodular anterior scleritis. Indomethacin 50 to 75 mg three times daily with food is the most commonly used agent and is effective in 50 to 70% of mild-to-moderate cases. Alternatives include ibuprofen 600 to 800 mg three times daily, naproxen 500 mg twice daily, or flurbiprofen. A course of 4 to 6 weeks is typical, followed by gradual tapering guided by clinical response. Patients must be counseled about gastrointestinal side effects; a proton pump inhibitor is recommended alongside NSAID therapy.
Important note: Topical NSAIDs alone (ketorolac eye drops, for example) do not provide adequate anti-inflammatory coverage for the sclera, which is too deep for meaningful topical drug penetration. They may reduce surface discomfort but do not treat the underlying scleral inflammation.
Oral Corticosteroids
Oral prednisone is used for moderate-to-severe non-necrotizing scleritis that does not respond adequately to NSAIDs within 2 to 4 weeks, and as initial therapy for severe disease. The standard starting dose is 1 mg/kg/day (typically 40 to 80 mg daily), maintained until inflammation is controlled, then tapered over 4 to 6 weeks. Gastric protection (PPI), calcium and vitamin D supplementation, and monitoring for steroid side effects (blood pressure, blood glucose, bone density) are standard adjuncts.
Subconjunctival Steroid Injection
Depot methylprednisolone or triamcinolone injected subconjunctivally can be effective for localized nodular anterior scleritis that has not responded to oral therapy. However, this approach is contraindicated in necrotizing scleritis. In necrotizing disease, the injection itself — placed into already-compromised scleral tissue — carries the risk of perforation at the injection site. The distinction between nodular (injection permissible) and necrotizing (injection forbidden) must be made before considering this route.
Treatment of Posterior Scleritis
Posterior scleritis typically responds well to systemic oral corticosteroids, often with dramatic improvement in pain, choroidal folds, and exudative detachment within days to weeks. It does not require a higher initial dose than other severe anterior subtypes, but the clinical response should be confirmed on repeat B-scan or OCT to ensure resolution of the choroidal thickening and subretinal fluid.
Treatment of Infectious Scleritis
Infectious scleritis requires pathogen-specific treatment. Herpes zoster scleritis requires systemic antivirals (valacyclovir 1g three times daily for 7 to 14 days). Bacterial scleritis requires culture-guided antibiotic therapy with appropriate topical and systemic agents. Importantly, SINS (surgically induced necrotizing scleritis) is immune-mediated and responds to systemic immunosuppression — not antibiotics. The misdiagnosis of SINS as infection is common and can delay life-saving immunosuppressive treatment.
Treatment — Immunosuppressive and Biologics
Indications for Systemic Immunosuppression
Immunosuppressive agents beyond corticosteroids are indicated when: (1) scleritis is the necrotizing subtype; (2) the patient is steroid-dependent (cannot taper below a dose associated with steroid side effects without disease flare); (3) the disease fails to respond to steroids alone; (4) the patient has a systemic autoimmune disease that itself requires immunosuppressive management; or (5) posterior scleritis with visual complications is present. The choice of agent is guided by systemic disease, organ toxicity profile, patient age, and severity.
Methotrexate
Methotrexate (MTX) is the most widely used conventional immunosuppressive for scleritis, particularly in the context of rheumatoid arthritis. The standard ophthalmologic dose is 15 to 25 mg per week, administered orally or subcutaneously (SC injections have more reliable bioavailability). Folic acid 1 mg daily is always co-prescribed to reduce mucosal and hematologic toxicity. Monthly monitoring of CBC and liver function tests is standard. MTX requires 4 to 6 weeks to reach meaningful anti-inflammatory effect, so it is often overlapped with corticosteroid therapy during induction.
Mycophenolate Mofetil
Mycophenolate mofetil (MMF, CellCept) at doses of 1 to 3 grams daily is increasingly preferred as an alternative to MTX, particularly for patients who cannot tolerate MTX or have MTX-relative contraindications (hepatic disease, significant pulmonary disease, or significant alcohol use). MMF is generally better tolerated with a lower risk of hepatotoxicity and pulmonary toxicity. Its onset of action is also 4 to 8 weeks.
Azathioprine
Azathioprine at 1 to 2.5 mg/kg/day is a third-line conventional immunosuppressive option. It is less commonly used for scleritis than MTX or MMF but has an established record in systemic autoimmune disease management. Thiopurine methyltransferase (TPMT) genotyping or enzymatic activity testing before starting is recommended to identify patients at risk for myelosuppression.
Cyclophosphamide
Cyclophosphamide is the most powerful agent in the conventional immunosuppressive arsenal and is reserved for the most severe cases — necrotizing anterior scleritis with GPA, or rapidly progressing necrotizing scleritis threatening globe integrity or vision. It can be administered as intravenous pulse therapy (e.g., 500 to 1000 mg/m² monthly) or as daily oral therapy (1 to 2 mg/kg/day). Toxicities are significant and include myelosuppression, hemorrhagic cystitis, increased malignancy risk, and gonadal toxicity. Mesna (uroprotection), hydration, and close hematologic monitoring are mandatory adjuncts.
Anti-TNF Biologics
Infliximab (IV infusion, 3 to 5 mg/kg at 0, 2, and 6 weeks, then every 8 weeks) is the most extensively studied biologic for refractory scleritis. Multiple case series and retrospective studies have documented excellent efficacy in necrotizing and non-necrotizing refractory scleritis. Infliximab's IV route allows rapid dose adjustment and delivers high peak drug levels, making it particularly attractive for acute severe presentations. Screening for latent tuberculosis (PPD or IGRA) and hepatitis B is required before initiating all anti-TNF therapy.
Adalimumab (SC 40 mg every 2 weeks) offers the convenience of self-administered subcutaneous injection and has similar efficacy to infliximab in published scleritis series. It is FDA-approved for non-infectious uveitis and has off-label application for scleritis that is increasingly supported by case series data.
Rituximab (Anti-CD20)
Rituximab, a monoclonal antibody that depletes B cells by targeting the CD20 antigen, has become a key agent for GPA-associated necrotizing scleritis and RA-associated refractory disease. It is given as IV infusions (typically 1000 mg on days 1 and 15, or 375 mg/m² weekly for 4 doses, the rheumatologic regimen). Published case series show high rates of disease remission in previously refractory necrotizing scleritis, with many patients achieving sustained remission after one or two courses. Rituximab is now considered first-line (with steroids) for GPA by ACR/EULAR guidelines, which directly benefits the ocular manifestations.
Other Biologics
Tocilizumab (anti-IL-6 receptor) has emerging evidence for scleritis, particularly in RA-associated disease where IL-6 is a key inflammatory mediator. Abatacept (anti-CTLA-4, T-cell co-stimulation blockade) has case report and series support for RA-associated scleritis refractory to MTX and anti-TNF agents.
Duration of Treatment
For most patients requiring immunosuppression, treatment duration is a minimum of 12 to 24 months of maintained remission before a cautious taper attempt. Necrotizing scleritis, especially in the context of systemic vasculitis, may require indefinitely sustained low-dose immunosuppression. Treatment decisions are best made in collaboration between the ophthalmologist and the rheumatologist or uveitis specialist managing the patient's systemic disease.
Episcleritis — Diagnosis and Management
Episcleritis deserves a dedicated section because it is far more common than scleritis, is frequently the presenting concern at the ophthalmologist's office, and yet is fundamentally different in prognosis and required treatment. Understanding episcleritis clearly helps patients and clinicians avoid both under-treatment (mistaking scleritis for episcleritis) and over-treatment (subjecting a patient with simple episcleritis to unnecessary systemic workup and immunosuppression).
What Episcleritis Is
Episcleritis is inflammation of the episcleral tissue — the thin, vascular, connective tissue plane lying immediately beneath the conjunctiva and overlying the sclera. The episclera is a distinct tissue layer from the sclera, and inflammation confined to it does not carry the structural threat or the systemic disease burden of true scleral inflammation. Episcleritis appears as a sectoral or diffuse area of redness, which is often bright red rather than the dark violaceous color of scleritis. The inflammation is superficial enough that episcleral vessels are mobile when probed and blanch on phenylephrine testing.
Types
Simple (diffuse) episcleritis is the more common form, presenting with a broad sector or the entire episclera becoming red and mildly uncomfortable. It is acutely onset, typically reaching peak redness within hours, and resolves spontaneously in 2 to 3 weeks in most cases.
Nodular episcleritis presents with a raised, movable, tender, inflamed nodule on the episcleral surface. Unlike scleritic nodules, these are soft and can be shifted slightly with a cotton-tipped applicator. Nodular episcleritis is more likely to recur and may be somewhat more persistent than the diffuse form, though it still carries an excellent prognosis.
Management
For mild episcleritis: Artificial tears and cool compresses are sufficient. The condition will resolve on its own. No systemic workup is required for a first episode of simple episcleritis without systemic symptoms.
For moderate or more symptomatic episcleritis: Topical NSAIDs (ketorolac tromethamine 0.4% four times daily) or mild topical steroids (fluorometholone 0.1% or loteprednol 0.5% — avoiding potent steroids such as prednisolone acetate 1% for episcleritis alone, as the long-term steroid risk outweighs a self-limiting condition) can reduce discomfort and shorten the episode. Oral NSAIDs (ibuprofen 400 to 600 mg three times daily for 1 to 2 weeks) are an option for more uncomfortable episodes.
For frequent recurrence: A systemic workup becomes appropriate when episodes recur more than three or four times per year, when nodular episcleritis is present, or when there are systemic symptoms suggesting inflammatory disease. Oral NSAID prophylaxis during recurrences is often sufficient for chronic episcleral disease without systemic cause.
Prognosis of Episcleritis
Recurrence affects approximately 50% of patients, but each recurrence is typically self-limiting. Visual loss from episcleritis alone is extremely rare. The prognosis is excellent. Patients should be counseled that recurrences do not indicate failure of treatment or a worsening systemic condition — they are a feature of the episcleral tissue's tendency to cycle through inflammatory episodes.
Complications
The risk and type of complications in scleritis are closely tied to the subtype. Diffuse anterior scleritis has low complication rates when treated appropriately. Necrotizing scleritis — particularly if undertreated or diagnosed late — carries risks that extend beyond the sclera to involve the cornea, the optic nerve, and in rare cases the structural integrity of the entire globe.
Corneal Complications
Sclerosing keratitis develops when scleritis extends to involve the limbus (the junction of the cornea and sclera). Progressive opacification of the peripheral cornea occurs, reducing the transparency of the cornea and potentially narrowing the clear optical zone. In severe cases, the opacification advances centrally and reduces vision significantly.
Peripheral ulcerative keratitis (PUK) is a particularly dangerous complication — an immune-mediated corneal melt at the limbus that can coexist with necrotizing scleritis. PUK is a true ophthalmic emergency. The combination of PUK and necrotizing scleritis is called corneal-scleral necrosis. Without aggressive systemic immunosuppression (cyclophosphamide is often required), the peripheral cornea can perforate within days. PUK in the context of RA or GPA is a known marker of life-threatening systemic vasculitis and mandates urgent rheumatologic co-management.
Glaucoma
Secondary glaucoma can arise from multiple mechanisms in scleritis: direct trabeculitis (inflammation of the trabecular meshwork impeding aqueous drainage), anterior synechiae (adhesions pulling the peripheral iris forward to block the drainage angle), or steroid-induced elevation of intraocular pressure from long-term corticosteroid therapy. Intraocular pressure should be monitored at every visit during active disease and throughout steroid treatment.
Cataract
Posterior subcapsular cataracts are a common consequence of long-term oral or topical corticosteroid therapy. Inflammatory cataracts can also develop from chronic uveal involvement in the context of associated uveitis. Monitoring with periodic lens examination is appropriate for patients on sustained corticosteroid regimens.
Cystoid Macular Edema
Posterior segment involvement — particularly in posterior scleritis or in severe anterior scleritis with choroidal involvement — can cause cystoid macular edema (CME), a fluid accumulation in the macular photoreceptor layer that blurs central vision. OCT readily detects CME and can guide adjustments in anti-inflammatory therapy.
Scleral Thinning and Staphyloma
Progressive or inadequately treated scleritis — particularly the necrotizing subtypes — leads to permanent scleral thinning. The thinned sclera can bulge outward under the pressure of the intraocular fluid, forming a dark-colored protrusion called a staphyloma, where the underlying pigmented uvea becomes visible through or protrudes through the attenuated scleral wall. Staphylomata are a permanent structural change and a marker of prior severe inflammation.
Globe Perforation
Spontaneous globe perforation is rare but represents the most catastrophic complication of scleritis. It occurs in the setting of advanced necrotizing anterior scleritis or scleromalacia perforans, where the scleral wall has been progressively destroyed. Any minor external trauma — even rubbing the eye — can be enough to perforate the globe when the scleral thickness is reduced to a fraction of its normal 0.5 to 1 mm. Emergency surgical intervention (scleral patch graft) and removal of all inflammatory stimuli are required.
Prognosis and Monitoring
Prognosis by Subtype
Diffuse anterior scleritis carries a generally favorable prognosis. The majority of patients achieve disease control with oral NSAIDs or a short course of oral corticosteroids. Visual prognosis is excellent in most cases, with few patients experiencing permanent structural damage when the disease is properly managed.
Nodular anterior scleritis tends to recur more frequently than the diffuse subtype — up to 50 to 60% of patients experience at least one recurrence — but visual outcomes are generally favorable. Subconjunctival steroid injection is often effective for stubborn nodules.
Necrotizing anterior scleritis with inflammation carries the most serious prognosis. Studies have shown that 25 to 40% of patients suffer significant visual loss without aggressive treatment. It is associated with the highest rates of systemic disease, particularly RA and GPA. Patients with this subtype require long-term monitoring and often sustained immunosuppressive therapy.
Scleromalacia perforans progresses slowly and silently. Because it is painless, damage can be extensive before the patient presents. Visual acuity is often preserved until structural instability of the globe develops. Regular monitoring even in the absence of symptoms is essential.
Posterior scleritis typically responds well to systemic corticosteroids and carries a good visual prognosis when recognized and treated. The exudative retinal detachments and choroidal folds resolve with appropriate anti-inflammatory therapy in most patients. The main risk is misdiagnosis — if treated as a tumor rather than inflammation, the opportunity for effective treatment is missed.
Monitoring Schedule
During active disease, slit-lamp examination should be performed at least monthly, and more frequently (every 1 to 2 weeks) during periods of disease escalation or treatment change. Intraocular pressure (IOP) should be checked at every visit. OCT imaging is recommended when posterior segment involvement is suspected or confirmed. Patients on long-term corticosteroids should undergo annual bone density (DEXA) scanning, regular blood glucose and blood pressure monitoring, and periodic lens examination for cataract formation.
When to Re-Escalate Treatment
Warning signs that require prompt reassessment and possible treatment intensification: any new visual symptoms; deepening or worsening pain; appearance of an avascular scleral patch; new corneal involvement; rising intraocular pressure refractory to topical agents; or worsening choroidal thickening or subretinal fluid on imaging. In patients with systemic disease, scleritis flares often parallel systemic disease activity; coordinated care between ophthalmology and rheumatology improves outcomes in both domains.
Key Research Papers
The following peer-reviewed publications form the evidence base for understanding and managing scleritis. All citations link to PubMed.
- Watson PG, Hayreh SS. "Scleritis and episcleritis." Br J Ophthalmol. 1976;60(3):163–191. PMID: 1268179 — The foundational classification system for scleritis and episcleritis; described the anatomical and clinical distinctions that remain standard today.
- Jabs DA, Mudun A, Dunn JP, Marsh MJ. "Episcleritis and scleritis: clinical features and treatment results." Am J Ophthalmol. 2000;130(4):469–476. PMID: 11024419 — Landmark retrospective series of 172 patients defining clinical characteristics, systemic disease associations, and treatment outcomes across scleritis subtypes.
- Foster CS, Sainz de la Maza M. The Sclera. Springer, 1994. — The authoritative textbook on scleritis; provides comprehensive classification, systemic associations, and historical treatment data.
- Sainz de la Maza M, Foster CS, Jabbur NS. "Scleritis associated with rheumatoid arthritis and with other systemic immune-mediated diseases." Ophthalmology. 1994;101(7):1281–1288 — Search PubMed — Describes the strong association between scleritis subtypes (especially necrotizing) and rheumatoid arthritis and vasculitic diseases.
- Raiji VR, Palestine AG, Parver DL, et al. "Scleritis and systemic disease association in a community-based referral practice." Am J Ophthalmol. 2009;148(6):946–950 — Search PubMed — Epidemiologic study demonstrating systemic disease frequency across scleritis subtypes; 38% of patients had associated systemic disease at diagnosis.
- Tuft SJ, Watson PG. "Progression of scleral disease." Ophthalmology. 1991;98(4):467–471 — Search PubMed — Long-term follow-up of necrotizing scleritis demonstrating progression, vision loss rates, and the importance of systemic immunosuppression.
- Smith JR, Mackensen F, Rosenbaum JT. "Therapy insight: scleritis and its relationship to systemic autoimmune disease." Nat Clin Pract Rheumatol. 2007;3(4):219–226 — Search PubMed — Comprehensive review of the mechanistic links between autoimmune vasculitis and scleritis, with treatment algorithm.
- Hatton MP, Durand ML, Bhatt A, et al. "Peripheral ulcerative keratitis and scleritis: a critical review." Int Ophthalmol Clin. 2002;42(1):89–99 — Search PubMed — Focused discussion of the combined corneal-scleral necrotizing syndrome, systemic disease associations, and treatment recommendations.
- Benson WE. "Posterior scleritis." Surv Ophthalmol. 1988;32(5):297–316 — Search PubMed — Seminal review of 65 cases of posterior scleritis; described the "T-sign" on B-scan and defined the clinical spectrum of this underdiagnosed entity.
- Cao JH, Oray M, Cocho L, Foster CS. "Rituximab in the treatment of refractory noninfectious scleritis." Am J Ophthalmol. 2016;164:22–28 — Search PubMed — Prospective study of rituximab for refractory scleritis showing significant improvement in disease activity and steroid-sparing effect.
- González-González LA, Molina-Prat N, Doctor P, et al. "Clinical features and presentation of posterior scleritis: a report of 31 cases." Ocul Immunol Inflamm. 2014;22(3):203–207 — Search PubMed — Large case series characterizing the clinical spectrum of posterior scleritis, diagnostic findings, and treatment response.
- Albini TA, Rao NA, Smith RE. "The diagnosis and management of anterior scleritis." Int Ophthalmol Clin. 2005;45(2):191–204 — Search PubMed — Practical review of stepwise diagnosis and treatment of anterior scleritis subtypes, including the phenylephrine test, systemic workup, and immunosuppressive algorithms.
Connections
- Ophthalmology
- Uveitis
- Conjunctivitis
- Blepharitis
- Dry Eye Disease
- Corneal Ulcer
- Orbital Cellulitis
- Rheumatoid Arthritis