Heparin-Induced Thrombocytopenia
Table of Contents
- Overview
- Types of HIT
- Pathophysiology
- Clinical Presentation
- Diagnosis — 4Ts Scoring System
- Management
- Special Situations
- Prognosis
- Key Research Papers
- Featured Videos
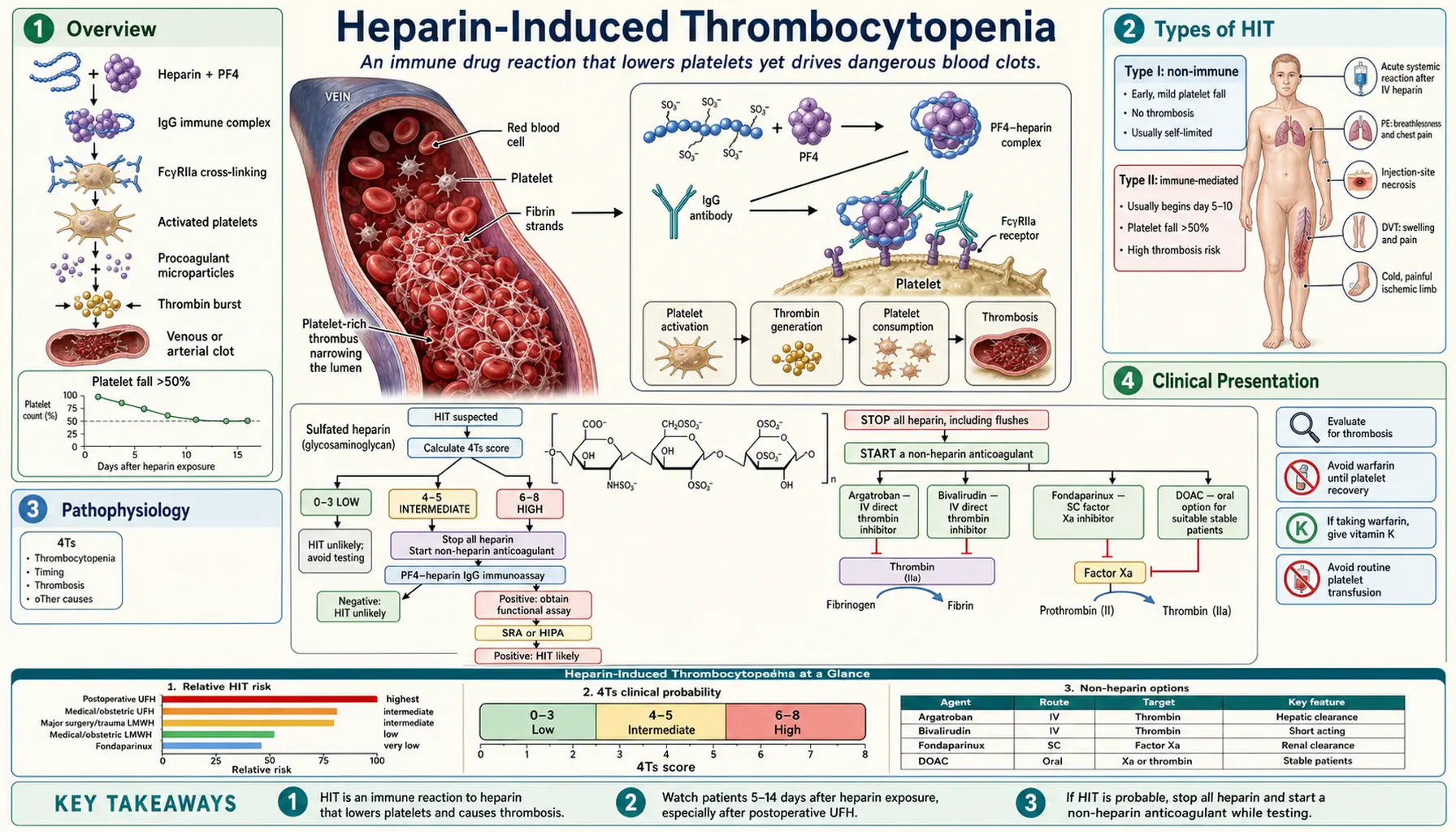
Overview
Heparin-induced thrombocytopenia (HIT) is an immune-mediated adverse drug reaction to heparin that produces a dangerous and counterintuitive clinical picture: a falling platelet count accompanied not by bleeding but by paradoxical, life-threatening thrombosis. This makes HIT the most dangerous iatrogenic coagulopathy encountered in hospital practice.
The incidence of clinically significant (Type II) HIT ranges from approximately 0.5% to 5% of heparin-exposed patients, varying by heparin type, dose, duration, and patient population. Surgical patients — particularly those undergoing cardiac or orthopedic procedures — face the highest risk. Despite the low absolute incidence, the sheer volume of heparin use in hospitals worldwide means HIT affects tens of thousands of patients annually.
The condition was first clearly described in 1958, when Roberts and colleagues noted paradoxical thrombosis in patients receiving heparin. For decades the mechanism remained unclear. The pivotal work of Theodore Warkentin, John Kelton, and Andreas Greinacher in the 1990s established that Type II HIT is driven by IgG antibodies targeting a complex of heparin bound to platelet factor 4 (PF4), a discovery that transformed clinical management.
The cardinal teaching point: HIT is a thrombotic disorder, not a bleeding disorder. Platelet transfusions — the instinctive response to a low platelet count — are contraindicated and can precipitate catastrophic thrombosis. Early recognition, immediate heparin cessation, and prompt substitution with a non-heparin anticoagulant are the cornerstones of survival.
Types of HIT
Type I HIT (Non-Immune)
Type I HIT is a mild, benign, and self-limiting phenomenon that occurs within the first 1–2 days of heparin initiation. It results from a direct, non-immunologic interaction between heparin molecules and platelet membranes, causing transient platelet sequestration.
- Platelet count rarely falls below 100,000/µL and typically normalizes spontaneously even if heparin is continued.
- No anti-PF4/heparin antibodies are detectable.
- No association with thrombosis.
- No specific treatment is required; the diagnosis is essentially one of exclusion after ruling out the more dangerous Type II form.
Type II HIT (Immune-Mediated — The Dangerous Form)
Type II HIT is the clinically significant entity. It is an antibody-mediated immune reaction with a characteristic 5–10 day window from heparin initiation to platelet decline. IgG antibodies form against the heparin-PF4 neoantigen, activate platelets and the coagulation cascade, and drive a hypercoagulable state that can produce devastating venous and arterial thromboses. Immediate intervention is mandatory.
Rapid-Onset HIT
Patients who have received heparin within the preceding 100 days may carry pre-formed circulating anti-PF4/heparin antibodies. Upon re-exposure, the thrombocytopenia and thrombosis can begin within hours rather than days, because no time is needed to generate a new antibody response. Clinicians must ask about recent heparin exposure in any patient who develops acute thrombocytopenia shortly after heparin administration.
Delayed HIT
A rare but underrecognized variant in which thrombocytopenia and thrombosis develop after heparin has already been discontinued — sometimes up to 3 weeks post-exposure. The antibodies persist after heparin is stopped, continue to activate platelets through low-level endogenous PF4, and can cause thrombosis even in the absence of ongoing heparin therapy. Delayed HIT should be considered in any patient presenting with unexplained thrombosis within weeks of a heparin course.
Pathophysiology
The pathophysiology of Type II HIT is a precisely defined immunologic cascade that explains both the thrombocytopenia and — paradoxically — the thrombosis. Understanding each step is essential for rational management.
Step 1: Heparin-PF4 Complex Formation
Platelet factor 4 (PF4) is a positively charged (+18 net charge) chemokine stored in platelet alpha-granules and released upon platelet activation. Heparin, a highly negatively charged glycosaminoglycan, electrostatically binds to PF4 and induces a conformational change, exposing a neoantigen not present on PF4 alone or heparin alone. This heparin-PF4 complex is immunogenic.
Step 2: IgG Antibody Formation
After approximately 5–7 days of antigen exposure, the immune system generates IgG antibodies (predominantly IgG1 subclass) directed against the heparin-PF4 neoantigen. This explains the 5–10 day delay between heparin initiation and platelet count decline in most patients without prior exposure.
Step 3: Platelet Activation and Amplification
IgG-heparin-PF4 immune complexes bind simultaneously to the antigen-specific Fab region and, via the Fc portion of the IgG, to FcγRIIa receptors on platelet surfaces. This crosslinking triggers explosive platelet activation, leading to:
- Platelet aggregation and consumption (explaining the thrombocytopenia).
- Release of procoagulant microparticles from platelet membranes — phosphatidylserine-rich surfaces that dramatically accelerate thrombin generation.
- Additional alpha-granule release, spilling more PF4 into the circulation — creating a self-amplifying cycle that perpetuates antibody formation and platelet activation.
Step 4: Monocyte and Endothelial Activation
HIT IgG complexes also bind FcγR on monocytes and vascular endothelial cells, triggering tissue factor expression. Tissue factor activates the extrinsic coagulation pathway, generating massive quantities of thrombin — the final common pathway to clot formation.
Result: The "White Clot Syndrome"
The net effect is paradoxical: platelet consumption drives the platelet count down (thrombocytopenia) while simultaneously the activated platelets, procoagulant microparticles, and tissue factor generate a profoundly prothrombotic state. The resulting thrombi are platelet-rich "white clots" — dense aggregates of activated platelets bound together by fibrin strands, morphologically distinct from the red fibrin-dominant thrombi of stasis or venous thromboembolism. This is why HIT is sometimes called white clot syndrome.
Clinical Presentation
Thrombocytopenia
The hallmark laboratory finding is a platelet count falling to less than 150,000/µL or by more than 50% from the pre-heparin baseline. In HIT the nadir typically ranges from 40,000 to 80,000/µL — low enough to be alarming but rarely low enough to cause spontaneous bleeding. A platelet nadir below 20,000/µL is unusual and should prompt consideration of alternative diagnoses such as immune thrombocytopenic purpura (ITP) or thrombotic thrombocytopenic purpura (TTP).
Timing
In patients without recent prior heparin exposure, platelet decline begins on days 5–10 after heparin initiation. In patients with heparin exposure within the prior 100 days, onset may be within hours (rapid-onset HIT). In delayed HIT, thrombocytopenia manifests after heparin discontinuation.
Thrombosis
Thrombosis occurs in approximately 50–75% of HIT cases and is the primary driver of morbidity and mortality. It may predate clinical recognition of thrombocytopenia.
- Venous thrombosis (most common): deep vein thrombosis (DVT) and pulmonary embolism (PE) are the most frequent manifestations.
- Arterial thrombosis (less common but more dramatic): acute limb ischemia from white clot occlusion of a major artery is a surgical emergency; stroke and myocardial infarction also occur. Arterial events are more limb- and life-threatening than venous events.
- Unusual sites: adrenal vein thrombosis causing hemorrhagic adrenal infarction and acute adrenal insufficiency; cerebral venous sinus thrombosis (can mimic other neurological emergencies).
Skin Necrosis at Heparin Injection Sites
Erythematous plaques or frank necrotic lesions at subcutaneous heparin injection sites are considered pathognomonic for HIT. They represent local microvascular thrombosis at sites of highest PF4-heparin concentration, and should immediately trigger HIT workup even if the platelet count has not yet fallen significantly.
Acute Systemic Reactions
Some patients develop an acute inflammatory or anaphylactoid reaction within minutes of receiving an intravenous heparin bolus: fever, chills, flushing, hypertension, tachycardia, dyspnea, or in severe cases, cardiac arrest. This represents massive, near- instantaneous platelet activation by pre-formed antibodies and is a medical emergency. Any such reaction after IV heparin should prompt immediate HIT evaluation.
Diagnosis — 4Ts Scoring System
Diagnosis of HIT requires integrating clinical probability with laboratory testing. The validated 4Ts scoring system provides a standardized pretest probability assessment before sending laboratory tests.
The 4Ts Score
Each "T" is scored 0, 1, or 2 points; maximum score is 8.
T1 — Thrombocytopenia Severity
- 2 points: Platelet fall >50% AND nadir ≥20,000/µL.
- 1 point: Platelet fall 30–50%, OR nadir 10,000–19,000/µL.
- 0 points: Platelet fall <30%, OR nadir <10,000/µL.
T2 — Timing of Platelet Fall
- 2 points: Clear onset days 5–10, OR ≤1 day with prior heparin exposure within 100 days.
- 1 point: Consistent with days 5–10 but not clear; OR onset >10 days; OR onset ≤1 day with prior heparin exposure >100 days ago.
- 0 points: Platelet fall <4 days without recent heparin exposure.
T3 — Thrombosis or Other Sequelae
- 2 points: New confirmed thrombosis; skin necrosis at heparin injection sites; acute systemic reaction after IV heparin bolus.
- 1 point: Progressive or recurrent thrombosis while on heparin; erythematous (non-necrotic) skin lesions at injection sites.
- 0 points: None.
T4 — oTher Causes of Thrombocytopenia
- 2 points: No other cause evident.
- 1 point: Possible other cause present.
- 0 points: Definite other cause present (e.g., sepsis, recent surgery, drug-induced thrombocytopenia from another agent).
Score Interpretation
- 6–8 points: High probability — approximately 50% prevalence of HIT. Treat empirically; confirm with laboratory testing.
- 4–5 points: Intermediate probability — approximately 10% prevalence. Laboratory testing required.
- 0–3 points: Low probability — <1% prevalence. HIT effectively excluded; investigate other causes.
Laboratory Testing
Two categories of tests are available:
- Anti-PF4/Heparin ELISA (immunoassay): High sensitivity (~95%) but lower specificity — detects anti-PF4 antibodies regardless of whether they actually activate platelets. Many patients form antibodies without developing clinical HIT. The optical density (OD) value matters: an OD >2.0 correlates much more strongly with functional platelet-activating antibodies and clinical HIT. A negative ELISA effectively rules out HIT. A positive ELISA requires clinical correlation or confirmatory functional testing.
- Serotonin Release Assay (SRA): The functional gold standard. Washed donor platelets are incubated with patient serum and therapeutic concentrations of heparin; release of [14C]-serotonin above 20% constitutes a positive result. Sensitivity ~95%, specificity ~99%. Complex methodology, radioactive substrate, and limited availability (few reference laboratories) restrict its routine use; results may take days.
- Heparin-Induced Platelet Activation (HIPA) assay: A European alternative functional assay using multiple donor platelet pools; comparable performance to SRA.
In practice, the ELISA is sent first for its rapid turnaround; clinical management decisions (heparin cessation, alternative anticoagulation) should not await laboratory confirmation in intermediate- or high-probability cases.
Management
Management of suspected or confirmed HIT follows a sequential, time-critical protocol. Every step matters; errors at any stage can be fatal.
Step 1 — Stop All Heparin Immediately
Discontinue all forms of heparin — unfractionated heparin (UFH), low- molecular-weight heparin (LMWH), heparin flushes used to maintain catheter patency, and heparin-coated catheters or devices. LMWH cannot substitute for UFH in HIT: LMWH cross-reacts with HIT antibodies in approximately 85–95% of cases and is contraindicated.
Step 2 — Start Non-Heparin Anticoagulation Immediately
The risk of thrombosis in untreated HIT is approximately 30–50% within 30 days, even after heparin cessation. Non-heparin anticoagulation must be initiated regardless of whether thrombosis has occurred.
- Argatroban (direct thrombin inhibitor): Hepatically metabolized — preferred in renal failure. Administered as a continuous IV infusion, titrated to aPTT. Important caveat: argatroban prolongs the PT/INR, complicating the transition to warfarin.
- Bivalirudin (direct thrombin inhibitor): Also used, particularly in the setting of cardiac surgery, percutaneous coronary intervention, or when argatroban is unavailable. Largely renally cleared.
- Fondaparinux (synthetic pentasaccharide, indirect factor Xa inhibitor): Very low cross-reactivity with HIT antibodies. Widely used off-label for HIT management because of ease of administration (once-daily subcutaneous injection) and robust safety data. Renally cleared — avoid in severe renal impairment (CrCl <30 mL/min).
- Danaparoid (anti-Xa mixture): Standard agent in Europe, limited availability elsewhere.
- Direct Oral Anticoagulants (DOACs — rivaroxaban, apixaban): Emerging evidence supports their use once the acute thrombotic phase has resolved and the platelet count has normalized. They offer convenience for long-term outpatient management.
Step 3 — No Platelet Transfusions
Platelet transfusions are contraindicated in active HIT. Because HIT is a thrombotic — not hemorrhagic — disorder, transfused platelets become additional substrate for FcγRIIa-mediated activation, generating more procoagulant microparticles and amplifying thrombin generation. The clinical metaphor is apt: transfusing platelets in HIT is "adding fuel to a fire." Reserve platelet transfusions for life-threatening bleeding combined with a platelet count below 20,000/µL, and only after weighing the substantial thrombotic risk.
Step 4 — Warfarin Caution: The Warfarin Paradox
Do not initiate warfarin until the platelet count has recovered to ≥150,000/µL. Warfarin in acute HIT depletes the natural anticoagulants protein C and protein S — which have shorter half-lives than the procoagulant factors II, VII, IX, and X — creating a transient hypercoagulable window. This can precipitate venous limb gangrene: catastrophic ischemic necrosis of an extremity despite (or because of) anticoagulation. This counterintuitive complication, termed the "warfarin paradox" by Warkentin, is among the most devastating iatrogenic injuries in hematology.
If warfarin was already initiated before HIT was recognized, administer vitamin K to reverse it promptly, then maintain therapeutic-dose non-heparin anticoagulation until platelet count normalizes before reintroducing warfarin with overlap.
Step 5 — Duration of Anticoagulation
Continue therapeutic anticoagulation for a minimum of 3 months. Transition to long-term oral anticoagulation (warfarin with INR 2–3, or a DOAC) once the acute phase resolves. Patients with HIT-associated thrombosis generally require the same duration as unprovoked VTE management, with individualized risk-benefit assessment.
Special Situations
HIT and Cardiac Surgery
Cardiopulmonary bypass requires large volumes of heparin and cannot proceed with active HIT or high-titer antibodies. Management options include:
- Delay surgery 3 months: Anti-PF4 antibodies typically wane to undetectable levels within 100 days, after which brief heparin re-exposure is possible under close monitoring.
- Bivalirudin for bypass: Short-acting direct thrombin inhibitor that can be used as the sole anticoagulant during bypass and clears rapidly, permitting hemostasis at circuit removal.
- Non-heparin circuit coating and alternative pump priming solutions are also under investigation.
HIT in Renal Failure
Argatroban (hepatically metabolized) is the preferred agent. Avoid fondaparinux (renally cleared, accumulates in CrCl <30 mL/min). Bivalirudin is partially renally cleared and requires dose adjustment. Danaparoid is also renally cleared and should be used with caution.
HIT in Pregnancy
Both warfarin and DOACs are contraindicated in pregnancy. Fondaparinux has the most accumulated safety data in HIT-complicating pregnancy and is the agent most clinicians use. Danaparoid is an alternative in regions where it is available. Clinical guidance is based on case series rather than randomized trials.
Future Heparin Re-Exposure
Once HIT antibodies have cleared (typically after ~100 days), brief heparin re-exposure for an unavoidable indication such as cardiac bypass surgery can be considered with extreme caution: confirm antibody negativity by ELISA immediately before exposure, use heparin only intraoperatively for the shortest effective duration, and restart non-heparin anticoagulation postoperatively immediately.
Vaccine-Induced Immune Thrombocytopenia and Thrombosis (VITT)
Following the rollout of adenovirus-vector COVID-19 vaccines (Oxford-AstraZeneca ChAdOx1 and Johnson & Johnson Ad26.COV2.S), a rare but severe syndrome emerged characterized by thrombocytopenia and thrombosis — particularly in unusual venous sites such as cerebral venous sinuses, splanchnic veins, and pulmonary arteries. The pathophysiology mirrors HIT precisely: spontaneously activating anti-PF4 antibodies that do not require heparin as a cofactor.
Management of VITT parallels HIT management: avoid heparin (contraindicated), use non-heparin anticoagulation, and administer intravenous immunoglobulin (IVIG) to compete with the pathogenic antibodies for FcγRIIa receptor binding. The HIT framework — developed over three decades — proved directly applicable to this vaccine-associated emergency, demonstrating the clinical importance of understanding the underlying pathophysiology.
Prognosis
With appropriate, timely management the prognosis of HIT has improved markedly over the past three decades, though the condition remains serious.
- Mortality: With appropriate treatment, overall mortality from HIT is approximately 5–10%, largely driven by thromboembolic complications — PE, stroke, and myocardial infarction — and by delays in diagnosis.
- Thrombosis risk: Without anticoagulation, 30–50% of HIT patients develop new or progressive thrombosis within 30 days of diagnosis, even after heparin has been stopped. This thrombotic risk — not the thrombocytopenia itself — defines the urgency of treatment.
- Limb amputation: Arterial thrombosis with limb ischemia leads to amputation in approximately 5–10% of cases where arterial events occur.
- Key message: Early recognition and rapid sequential action — stop heparin, start non-heparin anticoagulant, avoid platelet transfusions, defer warfarin — dramatically improves outcomes. The mnemonic holds true: thinking about HIT saves limbs and lives.
Monitoring Recommendations
Current ACC/AHA and ACCP guidelines recommend platelet count monitoring every 2–3 days for any patient receiving heparin for more than 4 days, as this is the window within which Type II HIT characteristically manifests. High-risk populations — postoperative cardiac and orthopedic patients — warrant particularly vigilant surveillance.
Awareness Is the Best Prevention
HIT is paradoxical in almost every respect: a drug given to prevent clots causes clots; a falling platelet count signals thrombotic, not hemorrhagic, danger; the intuitive treatment (platelets for low platelets; warfarin for thrombosis) can be catastrophic. Understanding this paradox is the foundation of prevention and survival.
Key Research Papers
- Warkentin TE et al. "Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin." N Engl J Med. 1995;332(20):1330–5. PMID: 7715641
- Greinacher A et al. "Laboratory diagnosis of heparin-induced thrombocytopenia and comparison of platelet aggregation test, heparin-induced platelet activation test, and platelet factor 4/heparin enzyme-linked immunosorbent assay." Transfusion. 1994;34(5):381–5. — Search PubMed
- Warkentin TE et al. "The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia." Ann Intern Med. 1997;127(9):804–12. — Search PubMed
- Cuker A et al. "Predictive value of the 4Ts scoring system for heparin-induced thrombocytopenia: a systematic review and meta-analysis." Blood. 2012;120(20):4160–7. PMID: 22990018
- Linkins LA et al. "Treatment and prevention of heparin-induced thrombocytopenia: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: ACCP guidelines." Chest. 2012;141(2 Suppl):e495S–e530S. PMID: 22315270
- Warkentin TE et al. "Heparin-induced thrombocytopenia: a prospective study." Am J Hematol. 1998;57(4):303–7. — Search PubMed
- Arepally GM, Ortel TL. "Clinical practice: heparin-induced thrombocytopenia." N Engl J Med. 2006;355(8):809–17. PMID: 16928996
- Greinacher A et al. "Fondaparinux in the treatment of acute heparin-induced thrombocytopenia." Thromb Haemost. 2010;103(1):119–25. — Search PubMed
- Schulman S et al. "Rivaroxaban for treatment of suspected or confirmed heparin-induced thrombocytopenia study." J Thromb Haemost. 2017;15(7):1336–1345. — Search PubMed
- Warkentin TE et al. "Platelet-activating anti-PF4/heparin antibodies associated with past exposure to heparin: the 'proximal cause' of HIT." J Thromb Haemost. 2003;1(12):2500–2. — Search PubMed
- Greinacher A et al. "Vaccine-induced immune thrombocytopenia and thrombosis." N Engl J Med. 2021;384(22):2092–2101. PMID: 33835769
- Warkentin TE. "Heparin-induced thrombocytopenia: diagnosis and management." Circulation. 2004;110(18):e454–8. — Search PubMed
Connections
- Hematology
- Thrombotic Thrombocytopenic Purpura
- Deep Vein Thrombosis
- Pulmonary Embolism
- Antiphospholipid Syndrome
- Cardiology Diseases
- Thrombocytopenia — HIT is one of the drug-induced immune causes of a low platelet count.