Acute Lymphoblastic Leukemia (ALL)
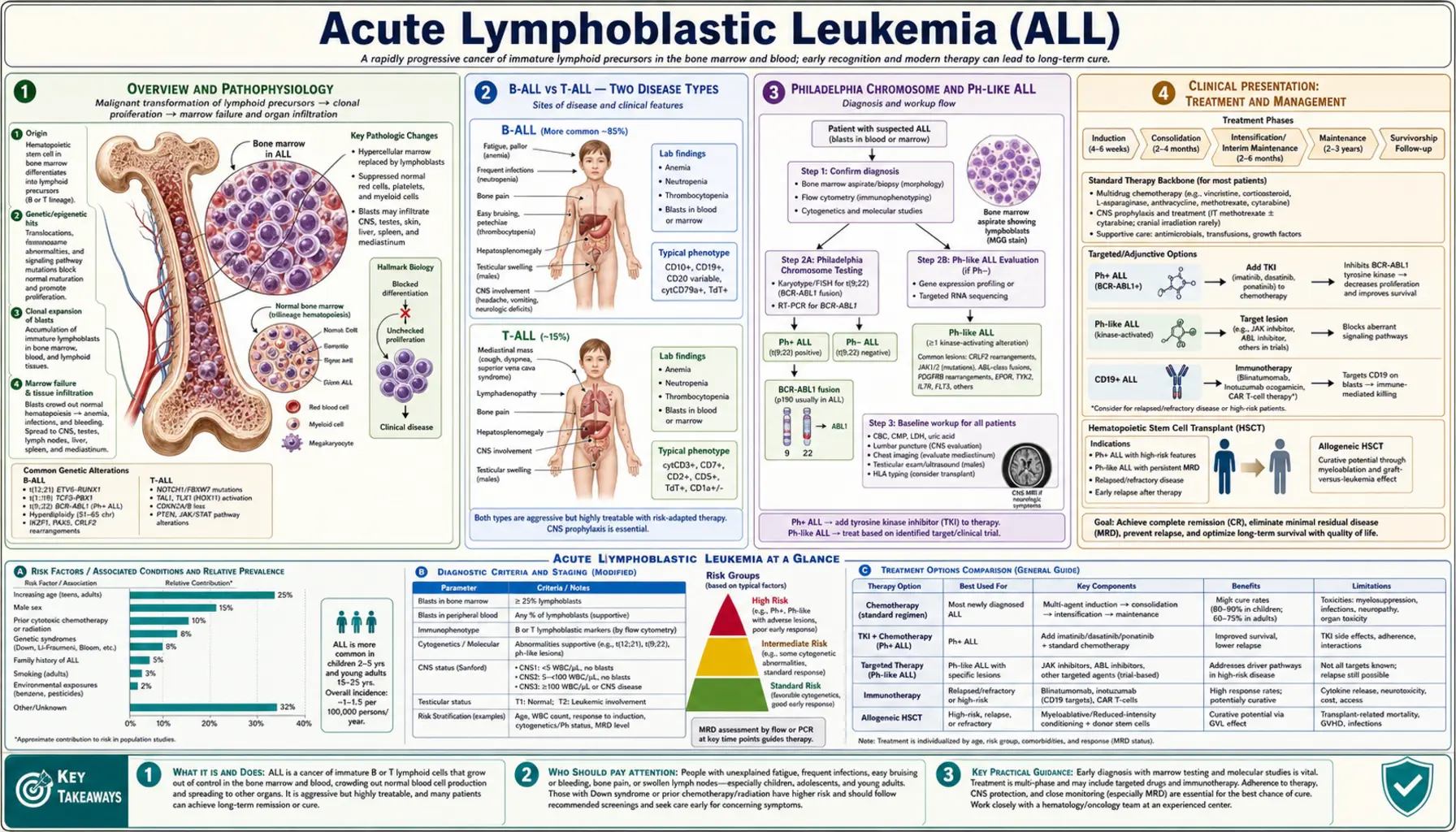
Acute Lymphoblastic Leukemia (ALL) is a fast-growing cancer of the blood and bone marrow. Abnormal white blood cells — called lymphoblasts — multiply out of control and crowd out the healthy blood cells your body needs. ALL is the most common cancer in children, accounting for about one in four of all childhood cancers, yet it also strikes adults, where it is harder to cure. The story of ALL is one of medicine's great success stories in children, and an ongoing challenge in adults.
Table of Contents
- Overview and Pathophysiology
- B-ALL vs T-ALL — Two Disease Types
- Philadelphia Chromosome and Ph-like ALL
- Clinical Presentation
- Diagnosis — Blood and Bone Marrow
- Risk Stratification and Prognosis
- Treatment — Pediatric Protocols (BFM/COG)
- Treatment — Adult ALL and TKI Therapy
- CNS Prophylaxis and Treatment
- Allogeneic Stem Cell Transplant
- Minimal Residual Disease (MRD)
- Relapsed and Refractory ALL
- Research Papers
- Connections
- Featured Videos
Overview and Pathophysiology
ALL begins when a single lymphoid progenitor cell — a very early type of white blood cell that normally matures into B cells, T cells, or natural killer cells — acquires a series of genetic mutations that block its ability to mature. Instead of growing up into a functional immune cell and stopping, the cell becomes stuck in an immature "blast" state and starts dividing without limits. These leukemic lymphoblasts multiply rapidly inside the bone marrow, where all blood cells are made.
As the blasts multiply they physically crowd out and suppress the production of normal red cells, normal white cells, and platelets. The result is pancytopenia — a dangerous shortage of all three blood cell types simultaneously. That shortage, rather than the blasts themselves, causes most of the symptoms a person feels.
About 6,500 new cases are diagnosed in the United States each year. The disease follows a bimodal age pattern: a large peak in children aged 2–5 years, then a smaller second peak in adults over 50. Children diagnosed at ages 1–9 with a low white cell count have a roughly 90–95% chance of cure with modern chemotherapy. Adults face a much steeper climb — 40–50% five-year survival overall, and worse in older patients.
The genetic events that drive ALL vary considerably. The most important categories are:
- Chromosomal translocations — pieces of two chromosomes swap places, creating fusion genes that hijack cell growth. The Philadelphia chromosome (BCR::ABL1), ETV6::RUNX1, TCF3::PBX1, and KMT2A rearrangements are the major examples.
- Changes in chromosome number (aneuploidy) — extra chromosomes (hyperdiploidy, more than 50 chromosomes total) generally predict a good outcome in children; too few chromosomes (hypodiploidy, fewer than 44) signal a very poor prognosis.
- Gene mutations — NOTCH1 pathway mutations occur in 55–60% of T-cell ALL; IKZF1 (Ikaros) deletions are a major adverse marker in adult B-ALL; JAK-STAT pathway mutations define a clinically important subgroup called Ph-like ALL.
Certain inherited conditions raise risk sharply. Children with Down syndrome (trisomy 21) have a 20-fold higher risk of ALL than the general population. Constitutional variants in IKZF1, Li-Fraumeni syndrome (germline TP53), and familial platelet disorder (RUNX1 mutations) are other recognized genetic predispositions. Prenatal origin has been demonstrated in some cases — the initiating chromosomal event can occur before birth, with additional mutations acquired in early childhood triggering overt leukemia.
B-ALL vs T-ALL — Two Disease Types
ALL is not one disease. It is divided into two broad subtypes based on which type of lymphoid cell has gone malignant: B-cell precursor ALL and T-cell ALL. They behave differently, respond to some treatments differently, and have different typical ages of onset.
B-Cell Precursor ALL (B-ALL)
B-ALL accounts for roughly 85% of all ALL cases. It arises from B-lymphoid progenitor cells that express the surface proteins CD19, CD22, and TdT (terminal deoxynucleotidyl transferase). Many also express CD10 (also called CALLA — common ALL antigen), which was historically used as a marker for "common" ALL and generally signals a better prognosis. B-ALL encompasses many genetic subtypes that behave quite differently from each other — ETV6::RUNX1-positive B-ALL in a child is nearly always curable, while KMT2A-rearranged infant ALL carries an extremely poor prognosis even with the most intensive available regimens.
T-Cell ALL (T-ALL)
T-ALL makes up about 15% of ALL cases. It arises from T-lymphoid progenitors in the thymus and expresses CD3, CD7, CD5, and TdT. T-ALL most often affects older children, adolescents, and young adults, and is more common in males. A hallmark clinical feature is a mediastinal (chest) mass — the leukemic cells accumulate in the thymus and can compress the trachea and the superior vena cava (the large vein draining the upper body into the heart). This can cause a life-threatening emergency.
Historically, T-ALL was thought to carry a worse prognosis than B-ALL. With modern intensified chemotherapy regimens, outcomes for T-ALL have improved substantially and now approach those of B-ALL in many centers. NOTCH1 pathway mutations, present in 55–60% of T-ALL, were initially expected to be a drug target; NOTCH1 inhibitors have been studied but not yet proven superior to standard chemotherapy. Nelarabine — a nucleoside analog specifically active in T-lymphoid cells — is an important agent for relapsed T-ALL.
Central nervous system (CNS) involvement is more common in T-ALL than B-ALL, so CNS-directed treatment must be adequate for both subtypes.
Philadelphia Chromosome and Ph-like ALL
Two genomically-defined subtypes of B-ALL have transformed treatment through targeted therapies: Philadelphia chromosome-positive ALL and Ph-like ALL. Understanding them matters because they are treated differently from other forms of ALL.
Philadelphia Chromosome-Positive ALL (Ph+ ALL)
The Philadelphia chromosome arises from a translocation between chromosomes 9 and 22 — written as t(9;22) — that creates the BCR::ABL1 fusion gene. This fusion encodes a constitutively active tyrosine kinase that drives uncontrolled cell proliferation. Ph+ ALL is present in about 25% of adult ALL cases but only about 3–5% of childhood ALL, and its frequency increases with age — by age 60 it may be present in nearly half of all B-ALL cases.
Before targeted therapy, Ph+ ALL had the worst prognosis of any ALL subtype — fewer than 10% of adults survived long-term. The introduction of BCR-ABL1 tyrosine kinase inhibitors (TKIs) transformed this disease. Adding imatinib, then dasatinib, then ponatinib to standard chemotherapy raised complete remission rates to 90%+ and five-year survival to 40–50% in adults. Dasatinib is often preferred because it crosses the blood-brain barrier and addresses CNS sanctuary disease more effectively. Ponatinib is the most potent TKI and is active against the resistant T315I mutation in ABL1; it is used both in relapsed/refractory Ph+ ALL and increasingly in frontline settings. The monoclonal antibody blinatumomab added to TKI+chemotherapy (the D-ALBA strategy) has further improved MRD negativity rates and may reduce the need for stem cell transplant in first remission for some patients.
Ph-like (BCR-ABL1-like) ALL
Ph-like ALL does not carry the BCR::ABL1 fusion but has a gene expression profile that closely resembles Ph+ ALL — hence the name. It accounts for 15–20% of B-ALL and is more common in adolescents and young adults. Outcomes are poor with standard chemotherapy. The defining biology is activation of kinase signaling pathways, and different genomic alterations activate these pathways in different patients:
- CRLF2 rearrangements (about 50% of Ph-like): often co-occurring with JAK1 or JAK2 mutations; responsive to JAK inhibitors such as ruxolitinib.
- ABL-class fusions (ABL1, ABL2, PDGFRA, PDGFRB, CSF1R): responsive to TKIs (imatinib, dasatinib, ponatinib) depending on which kinase is involved.
- EPOR rearrangements and other JAK-STAT activating alterations.
Clinical trials are actively investigating adding ruxolitinib (for CRLF2/JAK-mutant) or TKIs (for ABL-class fusions) to standard chemotherapy in Ph-like ALL. Identifying Ph-like ALL requires gene expression profiling or next-generation sequencing — it cannot be detected by routine cytogenetics or FISH alone.
KMT2A-Rearranged (MLL) ALL
Rearrangements of the KMT2A gene (formerly called MLL) on chromosome 11q23 define another biologically distinct subtype. The most common fusion partner is AFF1, creating the t(4;11) translocation. KMT2A-rearranged ALL is the dominant subtype in infants under 1 year old, where it carries a devastating prognosis — fewer than 30% survive long-term even with the most intensive chemotherapy and stem cell transplant. It is also present in some adult B-ALL cases and carries poor prognosis there as well. Novel agents targeting the menin-KMT2A interaction (menin inhibitors) are in clinical trials and have shown early promise in relapsed/refractory KMT2A-rearranged ALL.
Clinical Presentation
ALL typically develops over days to a few weeks. Symptoms arise primarily because the leukemic blasts crowding the bone marrow prevent the production of normal blood cells. Recognizing the constellation of signs can lead to faster diagnosis.
Symptoms of Bone Marrow Failure
- Fatigue, pallor, and shortness of breath — from anemia (low red cell count). Children may appear unusually pale and tire easily during play.
- Fever and recurrent infections — from neutropenia (low functional white cells). The fever may or may not mean active infection; sometimes it is driven by leukemia itself or by cytokines released by the blasts.
- Easy bruising, petechiae (pinpoint skin bleeding), nosebleeds, or gum bleeding — from thrombocytopenia (low platelet count). Small red or purple dots (petechiae) on the skin are a striking and important finding.
Lymph Node and Organ Enlargement
Lymphadenopathy — swollen lymph nodes, most often felt in the neck, armpits, or groin — is common. The liver and spleen may enlarge substantially as leukemic cells infiltrate them (hepatosplenomegaly), sometimes making the abdomen visibly distended. Bone pain, especially in the long bones of the legs, is a classic pediatric symptom — young children may simply refuse to walk or bear weight.
Mediastinal Mass (T-ALL Emergency)
In T-ALL, a large thymic mass in the chest can compress the trachea and the superior vena cava, causing facial swelling, neck vein distension, cough, and shortness of breath. This is an oncologic emergency. General anesthesia is extremely dangerous until the mass has been reduced, because lying flat and muscle relaxation can cause complete airway collapse. Diagnosis must be made from peripheral blood, lymph node biopsy under local anesthesia, or bone marrow biopsy rather than from a mediastinoscopy or open biopsy requiring general anesthesia.
CNS Involvement
ALL has a strong tendency to infiltrate the cerebrospinal fluid and leptomeninges (the membranes covering the brain and spinal cord). CNS involvement at diagnosis causes headache, nausea and vomiting, visual disturbances, and cranial nerve palsies (most commonly facial weakness or double vision). A lumbar puncture (spinal tap) to examine the CSF is mandatory at diagnosis. Retinal leukemia can sometimes be detected on fundoscopic examination.
Testicular Disease
In males, the testes can serve as a "sanctuary site" where leukemic cells hide from chemotherapy. Painless testicular swelling at diagnosis or relapse warrants biopsy to confirm leukemic infiltration. Testicular radiation or local treatment may be required.
Diagnosis — Blood and Bone Marrow
Diagnosing ALL requires more than finding abnormal cells in the blood. A complete workup establishes the lineage (B vs T), the stage of maturation arrest, the genetic subtype, and the presence of CNS disease — all of which directly determine treatment.
Complete Blood Count and Peripheral Smear
The CBC typically shows anemia, thrombocytopenia, and a white cell count that can range from very low (leukopenia) to extremely high (hyperleukocytosis above 100,000 per microliter). Lymphoblasts may be visible on the peripheral blood smear. They appear as cells with round or oval nuclei, fine chromatin, one or two small nucleoli, and very scant cytoplasm. Crucially, Auer rods are absent in ALL — their presence would indicate AML instead. TdT (terminal deoxynucleotidyl transferase) positivity helps confirm the blast is a lymphoid precursor.
Bone Marrow Biopsy and Aspirate
Bone marrow examination is required for definitive diagnosis and complete characterization. The marrow is almost always hypercellular (packed with cells) and usually contains more than 80–90% lymphoblasts, completely effacing the normal architecture. By convention, a diagnosis of ALL requires at least 20% blasts in the marrow, though most cases far exceed this threshold.
Immunophenotyping by Flow Cytometry
Flow cytometry examines the surface proteins (antigens) on each cell and is essential for defining ALL subtype. A B-cell precursor ALL immunophenotype shows positivity for CD19, CD22, and TdT, with variable CD10 (CALLA). T-ALL shows CD3, CD7, CD5, and TdT. Aberrant antigen expression — such as myeloid markers on lymphoblasts — does not change the diagnosis but may have prognostic implications. Mixed phenotype acute leukemia (MPAL), where blasts co-express both myeloid and lymphoid lineage markers beyond a defined threshold, is a separate diagnosis requiring its own treatment approach.
Cytogenetics and FISH
Standard cytogenetics (karyotype) evaluates the chromosomes under the microscope after stimulating cells to divide. This detects the Philadelphia chromosome, ETV6::RUNX1, KMT2A rearrangements, TCF3::PBX1, hyperdiploidy, and hypodiploidy. Fluorescence in situ hybridization (FISH) uses DNA probes to detect specific chromosomal changes with higher sensitivity even in non-dividing cells, and is used to confirm suspected translocations.
Next-Generation Sequencing (NGS) and Gene Expression Profiling
Comprehensive genomic panels detect mutations in IKZF1, NOTCH1, PTEN, PAX5, and RAS pathway genes; CRLF2 rearrangements; and ABL-class fusions that define Ph-like ALL. Gene expression profiling is needed to identify Ph-like ALL accurately. RNA sequencing is increasingly used to detect fusion transcripts. These tests are not optional extras — they directly guide risk stratification and treatment choices.
Lumbar Puncture and CSF Analysis
A lumbar puncture (spinal tap) is performed at diagnosis to examine the cerebrospinal fluid. CSF is classified as: CNS1 (no blasts detected), CNS2 (fewer than 5 white cells per microliter with blasts visible on cytospin), or CNS3 (5 or more white cells per microliter with blasts, or neurological signs of CNS leukemia). A traumatic lumbar puncture that introduces blood contaminated with peripheral blasts is treated as if CNS disease were present. CNS status determines how intensive the CNS-directed therapy needs to be.
Risk Stratification and Prognosis
One of the most important principles in ALL treatment is that not all patients need the same intensity of treatment. Treating everyone at the maximum dose would cause unacceptable toxicity in low-risk patients who would do fine with less. Risk stratification assigns each patient to a risk group based on factors known to predict treatment outcome.
Pediatric Risk Groups (NCI Criteria)
The National Cancer Institute (NCI) established two foundational risk groups based on age and white cell count at diagnosis:
- Standard Risk: Age 1–9 years AND white cell count below 50,000 per microliter. About 70% of childhood ALL patients fall here. Five-year overall survival exceeds 90–95% with modern regimens.
- High Risk: Age under 1 year OR age 10 or older OR white cell count at or above 50,000 per microliter. Any T-ALL is also classified as high risk regardless of other factors. Five-year survival is 75–85%, still excellent by historical standards but requiring more intensive therapy.
These categories are refined by cytogenetics and MRD response (see below). An initially standard-risk patient can be reclassified upward if they have adverse genetics or fail to clear their blasts quickly.
Favorable Genetic Features
- ETV6::RUNX1 fusion [t(12;21)]: present in about 25% of childhood B-ALL; confers excellent prognosis with 90%+ overall survival.
- Hyperdiploidy with more than 50 chromosomes: particularly favorable when trisomies of chromosomes 4, 10, and 17 are present simultaneously (the "good trisomies"); 25% of childhood ALL.
- Down syndrome ALL: paradoxically good prognosis in children despite the high incidence. However, these children are unusually sensitive to methotrexate toxicity, requiring dose modification.
Adverse Genetic Features
- Philadelphia chromosome (Ph+): most common adverse feature in adults; partially overcome by TKIs.
- Ph-like ALL: adverse in adolescents and adults; potentially targetable.
- KMT2A rearrangements: catastrophic in infants; poor in adults.
- Hypodiploidy (fewer than 44 chromosomes; near-haploid or low-hypodiploid): very poor prognosis; requires stem cell transplant in first remission in most centers.
- IKZF1 deletion: a deletion of the gene encoding Ikaros, a transcription factor critical for B-cell development; associated with poor prognosis in adult B-ALL, especially when co-occurring with CDKN2A/B deletion and PAX5 alteration ("IKZF1plus" profile).
Adult ALL Prognosis
Adult ALL is harder to cure for several reasons. Adults tolerate chemotherapy less well than children. A higher proportion of adult ALL carries adverse cytogenetics (especially Ph+). Many adults over 55 cannot receive full-intensity protocols due to organ function limitations. Despite these challenges, "pediatric-inspired" regimens — adult patients treated on protocols originally designed for older teenagers — have shown meaningfully better outcomes than traditional adult ALL protocols at academic centers, with some studies reporting 60–65% three-year overall survival in adults under 40.
Treatment — Pediatric Protocols (BFM/COG)
Pediatric ALL treatment is one of chemotherapy's greatest success stories. Over 60 years, cure rates have climbed from near zero to over 90% through a combination of multi-drug regimens, risk stratification, and intensive CNS-directed therapy. Treatment lasts 2 to 3 years total and is organized into phases.
Induction (Weeks 1–4)
The goal of induction is to get the leukemia into complete remission (no blasts detectable by standard microscopy in the marrow) within 4–6 weeks. The backbone drugs are:
- Vincristine — a vinca alkaloid that disrupts cell division by binding tubulin; given weekly by IV push; causes peripheral neuropathy over time.
- Dexamethasone or prednisone — a corticosteroid that directly kills lymphoblasts and reduces CNS leukemia; dexamethasone penetrates the CNS better and is preferred in many protocols for children.
- L-asparaginase (PEG-asparaginase) — an enzyme that depletes the amino acid asparagine from the blood. Lymphoblasts lack asparagine synthetase and cannot make their own supply; normal cells can upregulate this enzyme and survive. This makes asparaginase remarkably selective against leukemic cells. PEGylated asparaginase (Oncaspar) has a longer half-life and is given less frequently than the native enzyme. Erwinia asparaginase (Asparlas, Rylaze) is used for patients who develop hypersensitivity to PEG-asparaginase.
- Anthracycline (daunorubicin): added for high-risk patients to intensify the induction; not used in all standard-risk patients to reduce long-term cardiac toxicity.
Complete remission is achieved in over 95% of children after induction. The 5% or so who do not achieve CR after standard induction (induction failure) are among the highest-risk patients and almost always require stem cell transplant.
Consolidation and Intensification
Even in complete remission, residual leukemic cells that are undetectable by routine microscopy remain and will cause relapse without further treatment. Consolidation aims to eliminate these. Key elements include:
- High-dose systemic methotrexate — a folate antagonist given by intravenous infusion over 24 hours; penetrates the CNS at high doses; requires leucovorin rescue to prevent toxicity in normal cells; causes mucositis and kidney toxicity if clearance is impaired.
- 6-Mercaptopurine (6-MP) — daily oral thiopurine that interrupts purine synthesis; doses are adjusted based on TPMT/NUDT15 enzyme activity (genetic testing avoids dangerous toxicity in slow metabolizers).
- Cyclophosphamide and cytarabine — for high-risk patients in augmented BFM-style intensification blocks.
- Additional doses of asparaginase continue through consolidation; sustaining asparaginase activity is crucial for optimal outcomes.
Maintenance (2–3 Years)
Maintenance is the long phase that prevents relapse once the leukemia burden has been driven to its lowest level. The backbone is daily oral 6-MP plus weekly oral methotrexate, with monthly pulses of vincristine and a short steroid course. Maintenance is taken at home; the goal is to keep it going consistently — missed doses or underdosing significantly increase relapse risk. Thiopurine metabolite levels in the blood can be monitored to confirm adequate drug exposure.
Key Toxicities to Know
- L-Asparaginase: pancreatitis (usually self-limiting but can be severe); venous thrombosis (including cerebral sinus thrombosis — headache and neurological symptoms in a child on asparaginase must prompt imaging); hypersensitivity reactions; coagulopathy from reduced clotting factor synthesis.
- Methotrexate: mucositis (painful mouth sores), nephrotoxicity (impaired excretion prolongs toxicity), liver fibrosis (with prolonged maintenance), and rare acute neurotoxicity (stroke-like episodes, leukoencephalopathy).
- Vincristine: peripheral neuropathy (foot drop, weakness, loss of reflexes), constipation, and ileus; mostly reversible.
- Steroids (dexamethasone): avascular necrosis of the hip (osteonecrosis — especially in adolescents), hyperglycemia, mood and behavioral changes (can be severe in children), hypertension, and immune suppression.
- Anthracyclines: cumulative cardiac toxicity (cardiomyopathy); risk increases with higher total doses and is a concern for long-term survivors.
Treatment — Adult ALL and TKI Therapy
Treating ALL in adults has historically lagged far behind pediatric results. The same drugs cause more toxicity in older patients, dose-intensity cannot always be maintained, and the disease biology is less favorable on average. However, several advances have substantially changed the landscape for adult ALL.
Pediatric-Inspired Regimens in Adults
Traditional adult ALL protocols (HVAD, CALGB 8811) used lower cumulative doses of asparaginase and steroids to limit toxicity in adults. Observational data suggested that adults under 40 treated on pediatric-style protocols (such as COG AALL0434 for older adolescents) did better. Prospective studies have confirmed this: the CALGB 10403 trial treated adults aged 17–39 on a COG pediatric protocol, and reported a 3-year overall survival of 73% — dramatically better than historical controls. Pediatric-inspired regimens are now the standard of care for adults up to about age 40–45 at experienced centers.
Ph+ ALL — TKI-Based Frontline Treatment
The addition of TKIs to chemotherapy is the most transformative advance for adult ALL in the past two decades. For Ph+ ALL, the current standard approach varies by center and trial, but typically includes:
- Induction: steroids (dexamethasone) + a TKI (dasatinib most commonly; ponatinib increasingly in frontline) ± low-intensity chemotherapy. Some centers achieve 90%+ CR with a "chemotherapy-lite" or even chemotherapy-free induction using steroids + TKI + blinatumomab.
- Consolidation: standard multi-agent chemotherapy + continuous TKI.
- Blinatumomab integration: the D-ALBA trial (dasatinib + blinatumomab alternating) demonstrated that eliminating traditional intensive chemotherapy phases while achieving 97% MRD negativity is feasible in some Ph+ ALL patients, reducing toxicity without sacrificing efficacy.
- Allogeneic SCT in first CR for eligible patients, particularly if MRD-positive after consolidation.
Blinatumomab (Blincyto)
Blinatumomab is a bispecific T-cell engager (BiTE) — a very small antibody molecule with two binding arms, one attaching to CD19 on B-ALL blasts and the other to CD3 on cytotoxic T cells. It physically bridges the patient's own T cells to the leukemic cells, causing the T cells to kill them. It is given as a continuous 4-week IV infusion through a pump.
Blinatumomab is FDA-approved for relapsed/refractory B-ALL and for MRD-positive B-ALL in remission. In relapsed/refractory disease it achieves complete remission in about 40% of heavily pre-treated patients. Key side effects include cytokine release syndrome (CRS) — fever, low blood pressure, and oxygen requirements from immune activation — and neurological toxicity (confusion, seizures, encephalopathy) that typically resolves after brief drug interruption and dexamethasone. Strict monitoring protocols require immediate access to dexamethasone and expertise in managing these toxicities.
Inotuzumab Ozogamicin (Besylomab)
Inotuzumab ozogamicin is an antibody-drug conjugate: an anti-CD22 antibody chemically linked to calicheamicin, a potent DNA-damaging agent. CD22 is expressed on most B-ALL cells. The antibody delivers the toxin selectively into CD22-positive cells. In the INO-VATE trial, inotuzumab produced complete remission in 58% of patients with relapsed/refractory B-ALL versus 29% for standard chemotherapy. The major serious toxicity is hepatic sinusoidal obstruction syndrome (veno-occlusive disease, VOD) — a dangerous condition where small blood vessels in the liver are damaged. Risk is highest in patients who go on to receive a stem cell transplant after inotuzumab, and modified conditioning regimens are recommended to reduce VOD risk.
CNS Prophylaxis and Treatment
ALL is uniquely dangerous because of its tendency to infiltrate the brain and spinal cord lining. Before CNS-directed treatment was incorporated into ALL protocols in the 1960s and 1970s, virtually all children in remission eventually relapsed in the CNS. The leukemic blasts in the CSF are partially shielded from the full concentration of systemic chemotherapy by the blood-brain barrier — making CNS disease a "sanctuary site."
Why CNS Treatment Is Mandatory
Without any CNS-directed treatment, the relapse rate in the CNS would be 30–50% even in patients who achieve systemic complete remission. CNS relapse is extremely difficult to treat successfully and often heralds systemic relapse as well. Every ALL patient therefore receives CNS-directed therapy, regardless of whether CNS disease was detected at diagnosis.
Intrathecal Chemotherapy
Drugs injected directly into the spinal fluid via lumbar puncture reach the CNS at concentrations impossible to achieve with IV dosing. The standard drugs are:
- Methotrexate: given intrathecally during each phase of treatment (induction, consolidation, maintenance).
- Triple IT therapy (methotrexate + cytarabine + hydrocortisone): used in higher-risk situations and for CNS3 disease.
The number of intrathecal doses and the drugs used vary by protocol and CNS status at diagnosis. CNS3 patients receive intensified intrathecal treatment until the CSF clears, then continue prophylactic IT therapy through the rest of treatment.
High-Dose Systemic Methotrexate
At high intravenous doses (1–5 g/m² or higher), methotrexate crosses the blood-brain barrier at concentrations adequate to kill CNS leukemic cells. This is a cornerstone of CNS prophylaxis in modern protocols. It requires careful monitoring of drug levels and renal function, and timely leucovorin rescue to protect normal tissues.
Cranial Radiation
Cranial radiation was the original CNS prophylaxis, introduced in the 1970s and highly effective at preventing CNS relapse. However, long-term follow-up revealed serious consequences: cognitive impairment (IQ decline, learning disabilities, attention problems, memory deficits), neuroendocrine dysfunction (growth hormone deficiency, early puberty, hypothyroidism), increased risk of brain tumors, and stroke-like events decades later. Cranial radiation has been eliminated from most pediatric protocols and replaced by intrathecal + high-dose systemic methotrexate with equivalent efficacy and far less neurotoxicity. It is still used for overt CNS3 disease, some high-risk adult protocols, and certain transplant conditioning regimens (total body irradiation includes the brain).
Allogeneic Stem Cell Transplant
Allogeneic stem cell transplant (allo-SCT) uses high-dose chemotherapy — sometimes combined with total body irradiation — to destroy the patient's own bone marrow, followed by infusion of stem cells from a matched donor. The donor immune cells then engraft, produce new blood cells for the patient, and can attack any residual leukemia through the graft-versus-leukemia (GvL) effect. GvL is real but less potent in ALL than in AML.
When Is Transplant Needed in ALL?
In children, transplant is reserved for a narrow high-risk group because chemotherapy alone cures 90%+:
- Induction failure (not achieving CR after standard induction)
- Very high-risk cytogenetics (hypodiploidy; KMT2A-rearranged infant ALL)
- Ph+ ALL in some centers (though TKI-era data are beginning to support chemotherapy-only for MRD-negative patients)
- MRD-positive after consolidation in high-risk patients
In adults, indications for transplant in first complete remission are broader: Ph+ ALL, high-risk cytogenetics, MRD persistence after consolidation, and T-ALL with poor MRD response. Patients in second or later remission almost universally require transplant for any chance of durable cure.
Donor Sources
Matched sibling donor (a fully HLA-matched brother or sister) is the preferred source but is available only to about 25–30% of patients. Matched unrelated donor from a registry (National Marrow Donor Program / Be the Match) is used when no sibling is matched. Haploidentical (half-matched) transplant — usually from a parent or adult sibling — using post-transplant cyclophosphamide to prevent graft-versus-host disease has dramatically expanded access for patients who lack a fully matched donor, with outcomes now approaching matched unrelated donor in many series. Cord blood is an alternative, particularly for smaller patients where the cord unit cell dose is adequate.
Conditioning Regimens
TBI (total body irradiation) combined with cyclophosphamide or etoposide is the traditional conditioning regimen for lymphoid malignancies including ALL and is preferred in most centers. Busulfan-based conditioning is used when TBI is not available or not appropriate. Reduced-intensity conditioning extends transplant to older or less fit patients at the cost of somewhat higher relapse rates.
Graft-versus-Host Disease
The donor immune cells, while attacking leukemia, may also attack the patient's normal tissues in graft-versus-host disease (GVHD). Acute GVHD (skin rash, liver inflammation, gut cramping and diarrhea) occurs in the first 100 days; chronic GVHD resembles an autoimmune syndrome affecting skin, eyes, mouth, and lung and can persist for years. Management requires immunosuppression, which must be balanced against risking relapse.
Minimal Residual Disease (MRD)
Minimal residual disease (MRD) testing has become the single most important prognostic tool in ALL. It answers a question that standard bone marrow microscopy cannot: after a patient achieves "complete remission" and appears to have no visible blasts, how much leukemia actually remains?
The Problem MRD Solves
Standard morphology declares complete remission when fewer than 5% blasts are visible in the marrow. But that threshold still allows millions of leukemic cells to survive undetected. A patient with 4% morphologic blasts has far more residual leukemia than one with 0.001%. MRD tests detect one leukemic cell in 10,000 to 1,000,000 normal cells — orders of magnitude more sensitive than the microscope.
MRD Testing Methods
- Multiparameter flow cytometry: identifies leukemic cells by their unique immunophenotype (the specific combination of surface proteins that differs from any normal cell type). Sensitivity reaches 1 in 10,000 cells (10-4).
- PCR for clonal rearrangements or fusion transcripts: every leukemic cell carries a unique fingerprint in its immunoglobulin or T-cell receptor gene rearrangements, established at the time of malignant transformation. PCR amplifies this specific sequence to detect even rare leukemic cells. Sensitivity is 10-5 to 10-6. Also used to quantify BCR::ABL1 transcripts in Ph+ ALL.
- Next-generation sequencing (NGS-MRD; clonoSEQ): the most sensitive method available, reaching 10-6. FDA-cleared for ALL. Particularly useful when multiple leukemic clones are present.
MRD as a Treatment Guide
MRD results after induction and after consolidation are the most powerful predictors of relapse and survival in ALL. MRD negativity after induction — no detectable leukemia at 10-4 sensitivity — is associated with excellent long-term outcomes and may allow de-escalation of therapy in some low-risk patients. MRD positivity after consolidation signals high relapse risk and triggers treatment escalation: intensification with blinatumomab cycles, addition of novel agents, or proceeding to allogeneic stem cell transplant. Converting an MRD-positive patient to MRD-negative before transplant improves transplant outcomes.
Relapsed and Refractory ALL
When ALL returns after complete remission (relapse) or fails to respond to initial therapy (refractory disease), it becomes dramatically harder to cure. Second remissions can be achieved but are almost always shorter-lived than first remissions, and the leukemia has usually acquired resistance to drugs that worked before. Median overall survival for relapsed adult ALL treated with conventional salvage chemotherapy is 3–6 months. Novel agents have meaningfully extended this for some patients.
Blinatumomab in Relapsed ALL
Blinatumomab achieves complete remission in about 40% of patients with relapsed/refractory Ph-negative B-ALL and about 36% in Ph+ ALL (in combination with TKI). It is typically used as a bridge to stem cell transplant. Even patients who achieve only hematologic complete remission without MRD negativity benefit from transplant consolidation. Blinatumomab is also FDA-approved for MRD-positive B-ALL in remission — clearing MRD before it causes clinical relapse.
Inotuzumab Ozogamicin in Relapsed ALL
In the INO-VATE trial, inotuzumab achieved complete remission in 58% of patients compared to 29% with investigator's choice chemotherapy, and meaningfully extended overall survival. MRD negativity was achieved in 78% of inotuzumab responders. VOD risk after subsequent transplant requires careful attention to conditioning regimen selection.
CAR-T Cell Therapy — Tisagenlecleucel (Kymriah)
CAR-T cell therapy represents a fundamentally different approach. The patient's own T cells are collected by a process called leukapheresis, shipped to a manufacturing facility, and genetically engineered to express a chimeric antigen receptor (CAR) targeting CD19. After several weeks of manufacturing, the modified T cells are infused back into the patient, where they multiply and hunt down CD19-positive leukemic cells.
Tisagenlecleucel (Kymriah) is FDA-approved for pediatric and young adult ALL patients up to age 25 with B-ALL in second or later relapse or refractory disease. In the ELIANA trial, 81% of heavily pre-treated patients achieved remission — a response rate unachievable with any conventional chemotherapy. Durable remissions at three years occur in approximately 30–40% of patients. Key toxicities include:
- Cytokine release syndrome (CRS): fever, low blood pressure, oxygen requirements from massive immune activation; managed with tocilizumab (an IL-6 blocker) and corticosteroids.
- Immune effector cell-associated neurotoxicity syndrome (ICANS): confusion, aphasia, encephalopathy, and rarely cerebral edema; usually reversible with dexamethasone.
- Prolonged B-cell aplasia: CD19-targeting eliminates all B cells, causing hypogammaglobulinemia that requires monthly IVIG supplementation indefinitely.
CAR-T therapy is a single infusion, not a prolonged regimen. Manufacturing takes 3–4 weeks; bridging therapy during manufacturing is typically needed for patients with active disease.
Other Options in Relapsed ALL
- Nelarabine: a nucleoside analog converted intracellularly to ara-GTP, which is particularly toxic to T-lymphoid cells; used specifically in relapsed/refractory T-ALL; achieves CR in about 40%; can cause serious neurological toxicity.
- Ponatinib: the only TKI active against the T315I "gatekeeper" ABL mutation, which causes resistance to all first- and second-generation TKIs; used in relapsed Ph+ ALL with T315I, and increasingly in frontline Ph+ ALL.
- Venetoclax combinations: venetoclax (BCL-2 inhibitor) in combination with navitoclax, chemotherapy, or azacitidine is being investigated; early results show activity in ALL.
- Menin inhibitors (revumenib, ziftomenib): in clinical trials for KMT2A-rearranged and NPM1-mutant leukemias; early results show response rates of 30–40% in heavily pre-treated patients.
Research Papers
- Hunger SP, Mullighan CG (2015) — Acute Lymphoblastic Leukemia in Children. New England Journal of MedicineSearch PubMed. DOI link.
- Kantarjian H, et al. (2016) — Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. New England Journal of MedicineSearch PubMed. DOI link.
- Maury S, et al. (2016) — Addition of Dasatinib to Intensive Chemotherapy in Adolescents and Adults with Philadelphia Chromosome-Positive ALL. New England Journal of MedicineSearch PubMed. DOI link.
- Möricke A, et al. (2016) — Dexamethasone versus Prednisone in Induction Treatment of Pediatric ALL: Results of the European AIEOP-BFM ALL 2000 Study. Journal of Clinical OncologySearch PubMed. DOI link.
- Locatelli F, et al. (2021) — Tisagenlecleucel in Children and Young Adults with B-cell Lymphoblastic Leukemia. New England Journal of MedicineSearch PubMed. DOI link.
- Kantarjian H, et al. (2017) — Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. New England Journal of Medicine. PMID 27292104. DOI link.
- Bhojwani D, et al. (2015) — Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia. BloodSearch PubMed. DOI link.
- Pui CH, Yang JJ, Hunger SP, et al. (2015) — Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. Journal of Clinical OncologySearch PubMed. DOI link.
- Kantarjian HM, et al. (2017) — Blinatumomab versus Chemotherapy for Acute Lymphoblastic Leukemia (TOWER Study). New England Journal of MedicineSearch PubMed. DOI link.
- Stock W, et al. (2019) — What Determines the Outcomes for Adolescents and Young Adults with Acute Lymphoblastic Leukemia Treated on Cooperative Group Protocols? A Comparison of Children's Oncology Group and Cancer and Leukemia Group B Studies (CALGB 10403). Journal of Clinical OncologySearch PubMed. DOI link.
Search PubMed for More ALL Research
- Acute lymphoblastic leukemia treatment adults
- ALL Philadelphia chromosome TKI therapy
- CAR-T therapy ALL tisagenlecleucel
- Minimal residual disease ALL prognosis
Connections
- Hematology
- Acute Myeloid Leukemia
- Chronic Lymphocytic Leukemia
- Non-Hodgkin Lymphoma
- Hodgkin Lymphoma
- Myelodysplastic Syndrome
- Aplastic Anemia
- Thrombocytopenia
- Multiple Myeloma
- Leukemia — ALL is the acute lymphoid form of leukemia.