Tay-Sachs Disease
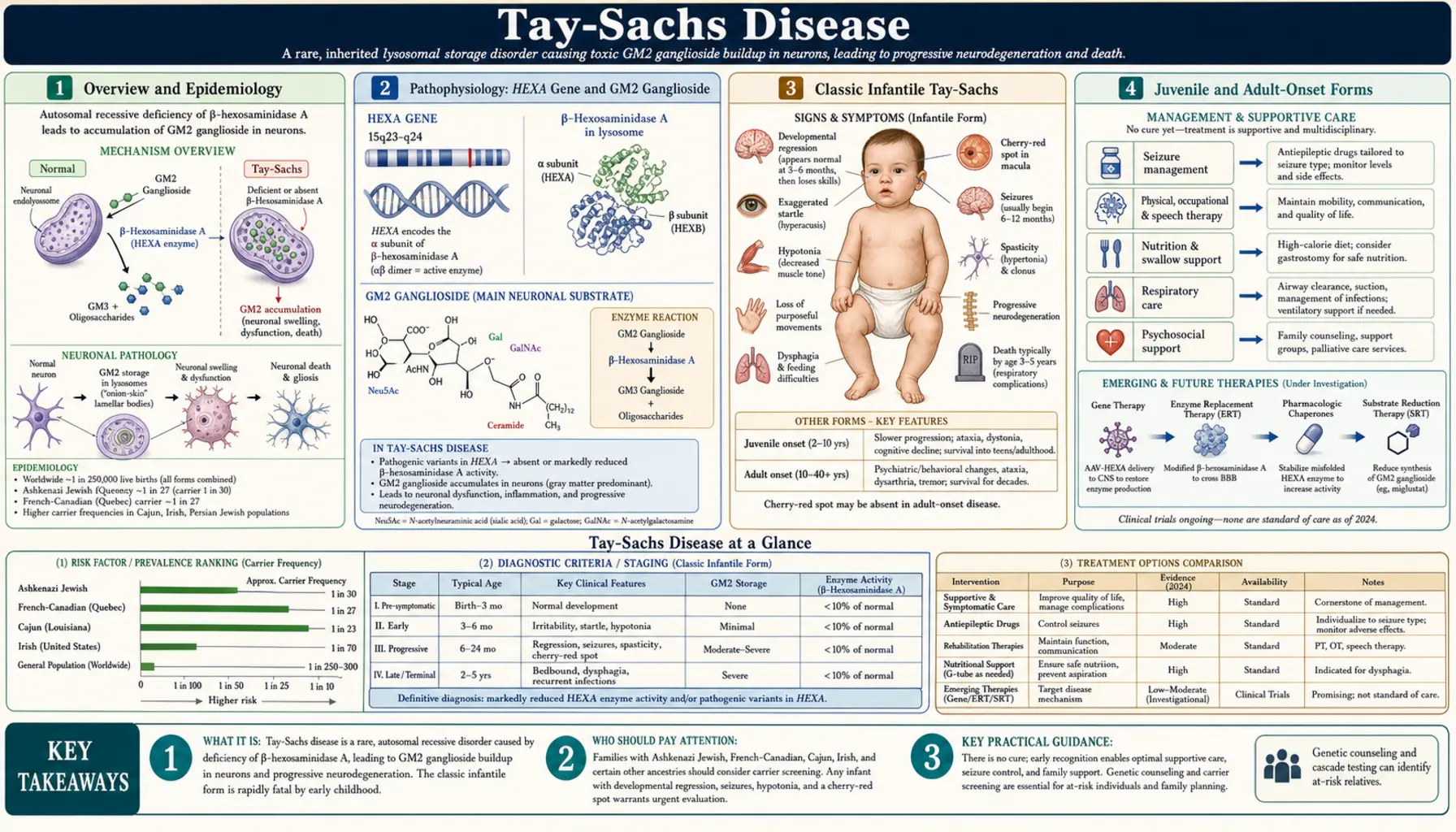
Overview and Epidemiology
Tay-Sachs disease is a fatal autosomal recessive lysosomal storage disorder caused by mutations in the HEXA gene on chromosome 15q23, which encodes the alpha-subunit of beta-hexosaminidase A (Hex-A). Without functional Hex-A, the ganglioside GM2 accumulates progressively within neuronal lysosomes, leading to relentless neurodegeneration and, in the classic infantile form, death before age five. The disease was independently described in the 1880s by British ophthalmologist Warren Tay, who noted the characteristic cherry-red spot on fundoscopy in 1881, and by American neurologist Bernard Sachs, who in 1887 described the full clinical and pathological syndrome — then called "amaurotic familial idiocy" — recognizing its familial pattern and Jewish ancestry clustering.
In the general population, carrier frequency is approximately 1 in 300. Among Ashkenazi Jews, the carrier rate rises dramatically to approximately 1 in 30 — roughly ten times higher — reflecting a founder effect from a small ancestral population. Elevated carrier frequencies are also documented in French Canadians of Quebec, Cajun communities of Louisiana, and individuals of Irish (Celtic) descent. In populations where targeted carrier screening programs have been implemented, particularly the Ashkenazi Jewish community beginning in the 1970s, the incidence of the devastating infantile form has been reduced by more than 90% — one of the great successes of population genetic screening in medicine.
Three clinical forms are recognized based on the amount of residual Hex-A activity and the age of onset: classic infantile (the most severe and most common), juvenile (subacute), and adult or late-onset Tay-Sachs (LOTS). All share the same underlying enzyme deficiency, but the degree of residual enzyme activity correlates inversely with disease severity and inversely with age of onset. The disease remains without an approved disease-modifying treatment, though gene therapy clinical trials are underway.
Pathophysiology: HEXA Gene and GM2 Ganglioside
The HEXA gene spans approximately 35 kilobases on chromosome 15q23 and encodes the 529-amino-acid alpha-subunit of beta-hexosaminidase A. Hex-A is an alpha-beta heterodimer: two subunits encoded by distinct genes (HEXA for alpha, HEXB for beta) assemble in the endoplasmic reticulum, traffic through the Golgi apparatus, and are delivered to the lysosome via the mannose-6-phosphate receptor pathway. Within the lysosome, Hex-A cleaves the terminal N-acetylgalactosamine residue from GM2 ganglioside (monosialodihexosylganglioside GM2), a glycolipid that is a normal structural component of neuronal cell membranes and is continuously generated during membrane turnover. This cleavage step is an obligate part of the GM2 catabolism pathway; without it, GM2 has no alternative route for degradation and accumulates irreversibly.
Three isoforms of beta-hexosaminidase exist in human tissues: Hex-A (alpha-beta heterodimer, active on GM2 ganglioside), Hex-B (beta-beta homodimer, active on other substrates but not GM2 ganglioside), and Hex-S (alpha-alpha homodimer, unstable and minimally active). In Tay-Sachs disease, the alpha-subunit is non-functional; Hex-B activity is therefore preserved and often elevated (since the beta-subunit continues to be produced and forms beta-beta homodimers). This distinction is diagnostically important: total hexosaminidase activity (Hex-A + Hex-B) may be normal or even elevated in Tay-Sachs, while the Hex-A fraction specifically is absent or severely reduced. The ratio of Hex-A to total hexosaminidase is the key diagnostic metric.
GM2 ganglioside accumulation begins prenatally in neurons and accelerates after birth as neuronal activity and membrane turnover increase. The ganglioside accumulates within lysosomes, causing progressive lysosomal swelling, disruption of axonal transport, mitochondrial dysfunction, activation of apoptotic pathways, and ultimately neuronal death. The process is highly selective for neurons — particularly large neurons such as motor neurons and the ganglion cells of the retina — because neuronal membranes are exceptionally rich in gangliosides compared to other cell types. The liver, spleen, and other visceral organs are largely spared, in contrast to Gaucher or Niemann-Pick disease, because those organs' macrophages generate far less GM2 substrate.
Related Disorders: Sandhoff and AB-Variant
Three distinct inherited disorders share the same pathological endpoint — lysosomal GM2 ganglioside accumulation causing neurodegeneration — but arise from mutations in three different genes. Together they are called the GM2 gangliosidoses, and distinguishing between them requires enzyme activity profiling rather than clinical criteria alone, because the infantile forms are clinically indistinguishable.
Sandhoff disease (also called GM2 gangliosidosis type II, or Hexosaminidase A and B deficiency) is caused by loss-of-function mutations in the HEXB gene, which encodes the beta-subunit shared by both Hex-A and Hex-B. Because Hex-A requires a functional beta-subunit to form its alpha-beta heterodimer, and Hex-B is itself a beta-beta homodimer, Sandhoff disease abolishes activity of both Hex-A and Hex-B simultaneously. Total hexosaminidase activity is essentially absent. Clinically, infantile Sandhoff disease is nearly identical to infantile Tay-Sachs — cherry-red spot, acoustic hyperekplexia, progressive neurodegeneration, death by age four. However, Sandhoff disease additionally causes mild accumulation of globoside (another hexosaminidase substrate) in visceral organs, occasionally producing mild hepatosplenomegaly — a finding absent in Tay-Sachs. Sandhoff is not enriched in the Ashkenazi Jewish population; its carrier frequency is uniform across ethnic groups at approximately 1 in 300.
AB-variant GM2 gangliosidosis is the rarest of the three, caused by mutations in the GM2A gene encoding the GM2 activator protein (GM2AP). GM2AP is a small lipid-transfer protein that binds the GM2 ganglioside substrate and presents it to Hex-A in a form the enzyme can process — Hex-A cannot efficiently cleave GM2 from intact ganglioside micelles without this activator. In the AB-variant, Hex-A enzyme activity is measurably normal when assayed with the synthetic substrate used in routine laboratory testing (which does not require GM2AP), but the enzyme cannot act on natural GM2 ganglioside in vivo. This creates a diagnostic trap: enzyme assay is falsely normal; only specialized activator protein assays, GM2A gene sequencing, or thin-layer chromatography of stored lipid can identify the correct diagnosis.
Classic Infantile Tay-Sachs
The classic infantile form is the most common and most severe presentation of Tay-Sachs disease. Affected infants are born after an uncomplicated pregnancy and appear entirely normal at birth, passing newborn screening and early developmental milestones. The first parental concern typically arises between three and six months of age, when the child seems to stop progressing or parents notice subtle regression — the baby who was beginning to reach for objects loses interest, the social smile becomes less frequent, and early motor development plateaus.
The exaggerated startle response to sound (acoustic hyperekplexia) is among the earliest and most pathognomonic clinical signs, often recognizable before other neurological abnormalities become obvious. An affected infant will startle with marked, exaggerated whole-body stiffening and extension in response to sudden loud noises — a response far greater than the normal Moro response and one that does not habituate with repeated stimulation (unlike the normal startle, which diminishes). Parents frequently notice this first as an unusually intense reaction to sounds in the home environment.
The cherry-red spot on fundoscopy is a classic finding, typically visible between six and twelve months. It arises because GM2 ganglioside deposits in the cytoplasm of retinal ganglion cells (which are densest at the macula) make the surrounding retina appear pale white or gray. The central macula, which normally appears slightly reddish due to its rich vascular supply and thin ganglion cell layer, stands out by contrast as a vivid red spot surrounded by pallid retina. While highly associated with Tay-Sachs disease, the cherry-red spot is not exclusive to it — it is also seen in Niemann-Pick disease types A and C, GM1 gangliosidosis, metachromatic leukodystrophy, Farber disease, and sialidosis, and rarely in central retinal artery occlusion.
The clinical course after six months follows a stereotyped, relentless trajectory of neurological decline. Hypotonia gives way to increasing spasticity; developmental milestones that were achieved are lost (developmental regression); seizures emerge, typically between twelve and eighteen months, initially as infantile spasms or myoclonic jerks and evolving to multiple refractory seizure types. Vision loss progresses to blindness from retinal ganglion cell destruction and optic atrophy. Cortical deafness ensues despite intact cochlear function. Decorticate and decerebrate posturing develop. By the third to fourth year, most children are in a vegetative state, requiring tube feeding and mechanical ventilation. Death typically occurs between two and five years of age, most often from respiratory failure, aspiration pneumonia, or intercurrent infection. There is no hepatosplenomegaly — the viscera are spared.
Juvenile and Adult-Onset Forms
Juvenile (subacute) Tay-Sachs disease presents in children between approximately two and ten years of age and follows a slower but ultimately fatal course. Unlike the classic infantile form, early development is normal, and the condition often comes to attention through school difficulties, clumsiness, or behavioral changes. Cerebellar ataxia and dysarthria are typically the earliest neurological signs. Cognitive decline and learning difficulties progress over months to years. Seizures occur in most patients but may not appear until years after onset. Visual deterioration is present but sometimes milder than in the infantile form; the cherry-red spot may be absent or less vivid. Death typically occurs in the teens or early twenties.
Adult or late-onset Tay-Sachs disease (LOTS) is a clinically distinct and widely underrecognized syndrome that can present deceptively as psychiatric illness or motor neuron disease. Onset typically occurs in the twenties or thirties. Patients retain significant residual Hex-A activity (higher than infantile or juvenile patients) and survive into adulthood, often for decades, but with progressive disability.
The neurological picture in adult Tay-Sachs is dominated by two overlapping phenotypes. The first is a progressive lower motor neuron disorder resembling spinal muscular atrophy: proximal limb weakness, fasciculations, reduced reflexes, and muscle atrophy from anterior horn cell degeneration. Upper motor neuron signs (spasticity, hyperreflexia) may coexist, giving a mixed upper and lower motor neuron picture that can superficially mimic amyotrophic lateral sclerosis (ALS). Cerebellar signs — ataxia, dysarthria, dysmetria — are frequently present. The second phenotype is psychiatric: adult Tay-Sachs frequently presents with psychosis, mania, depression, or a schizophrenia-like syndrome as the earliest manifestation, sometimes years before neurological signs appear. Because the psychiatric features are prominent and the diagnosis is rarely suspected in adults, patients with adult Tay-Sachs are routinely misdiagnosed with schizophrenia or bipolar disorder for years or even decades. Any patient with treatment-resistant psychosis combined with cerebellar signs, lower motor neuron features, or a family history suggesting storage disease should prompt Hex-A assay. Unlike the infantile form, adult Tay-Sachs does not typically cause cherry-red spot or epilepsy as prominent features.
Diagnosis: Enzyme Assay and Genetic Testing
The diagnostic cornerstone of Tay-Sachs disease is measurement of beta-hexosaminidase A (Hex-A) enzyme activity in peripheral blood leukocytes or serum. In affected individuals, Hex-A activity is markedly reduced or absent while total hexosaminidase activity (Hex-A + Hex-B) is preserved or even elevated, reflecting intact Hex-B (beta-beta homodimer). The critical metric is the ratio of Hex-A to total hexosaminidase; values below approximately 5–10% of normal are diagnostic of Tay-Sachs disease. Carriers typically show intermediate ratios of 40–60% of normal.
An important technical nuance is that serum assays are unreliable during pregnancy. Pregnancy physiologically elevates certain hexosaminidase isoforms (including an intestinal-derived Hex-B-like isozyme that co-elutes with Hex-A in heat-inactivation assays), leading to falsely elevated Hex-A ratios and potential misclassification of carriers as non-carriers or of affected individuals as carriers. Pregnant women suspected of being carriers or who have an affected fetus must be tested using the leukocyte (WBC) enzyme assay, which is not subject to this artifact. The heat-stability method separates Hex-A (heat-labile at 50°C) from Hex-B (heat-stable) and is used in routine serum testing; leukocyte assay using the fluorogenic substrate 4-methylumbelliferyl-beta-N-acetylglucosaminide-6-sulphate (MUGS), which is specifically cleaved by Hex-A but not Hex-B, is preferred for diagnostic and prenatal confirmation.
Molecular genetic testing of the HEXA gene complements enzyme assays. More than 150 pathogenic variants have been identified. In the Ashkenazi Jewish population, two mutations account for approximately 93% of pathogenic alleles: the c.1274_1277dup (4-base pair insertion in exon 11) causing a frameshift and premature stop codon — associated with the infantile form — and c.805G>A (p.Gly269Ser, G269S) associated with adult/chronic Tay-Sachs. In French Canadians of Quebec, a large deletion affecting intron 7 is the most common pathogenic allele. For prenatal diagnosis, chorionic villus sampling (CVS) at 10–12 weeks or amniocentesis at 15–18 weeks allows enzyme assay and/or mutation analysis on fetal tissue when both parents are confirmed carriers. Preimplantation genetic testing (PGT) is an option for couples undergoing IVF.
Neuroimaging and Electrophysiology
Brain MRI findings in infantile Tay-Sachs have a characteristic pattern that supports the clinical diagnosis when enzyme activity is not immediately available. In the first year of life, T2 and FLAIR sequences demonstrate increased signal intensity in the thalami — particularly the pulvinar and mediodorsal nuclei — and the basal ganglia (caudate and putamen). This "thalamic signal abnormality" or hyperintense thalami on T2-weighted imaging is highly characteristic, arising from the heavy neuronal density and GM2 accumulation in these structures. The cerebral cortex initially appears normal, but as disease progresses, diffuse cerebral atrophy becomes prominent along with progressive white matter signal changes (hypomyelination followed by demyelination). In later stages, severe cortical atrophy, thinning of the corpus callosum, and cerebellar atrophy are visible. The thalamic T2 hyperintensity stands in contrast to Sandhoff disease, where thalamic changes may be less prominent.
In adult Tay-Sachs disease, brain MRI shows cerebellar atrophy — often the most striking finding — with variable degrees of cerebral atrophy, white matter T2 signal changes in cerebellar peduncles and brainstem, and sometimes anterior horn cell loss detectable as T2 signal changes in the spinal cord anterior horns. MRI of the spine may demonstrate cord atrophy at cervical and thoracic levels corresponding to motor neuron degeneration. These findings, though non-specific individually, in combination with the clinical presentation of motor neuron disease plus psychiatric symptoms, should prompt immediate enzyme activity testing.
Electroencephalography (EEG) in infantile Tay-Sachs shows characteristic abnormalities. Early in the disease, background activity may be relatively preserved, but as seizures emerge, multifocal spikes and spike-wave complexes appear. Infantile spasms, when present, are accompanied by the hypsarrhythmia pattern on EEG. Progressive EEG disorganization parallels clinical deterioration. In juvenile Tay-Sachs, EEG may show progressive slowing with multifocal discharges; photoparoxysmal responses (light-induced spike-wave discharges) occur in some patients and reflect occipital cortex hyperexcitability. Electromyography (EMG) and nerve conduction studies in adult Tay-Sachs document the lower motor neuron pathology: reduced motor unit recruitment, fibrillation potentials, and fasciculation potentials in limb muscles, with normal or near-normal nerve conduction velocities — distinguishing the anterior horn cell degeneration from peripheral neuropathy.
Treatment: Symptomatic and Emerging Therapies
No disease-modifying therapy is currently approved for Tay-Sachs disease, and clinical management remains supportive and symptomatic. For families of children with infantile Tay-Sachs, honest and compassionate communication about prognosis, integration of palliative care, and meticulous attention to quality of life are the foundations of management.
Seizure management is central to the care of infantile and juvenile patients. Seizure control is often partial at best but can improve comfort and reduce distress. Infantile spasms may respond to ACTH, vigabatrin, or the ketogenic diet. As the disease progresses, multiple antiepileptic drugs (AEDs) are typically required; combinations including valproate, levetiracetam, clonazepam, and clobazam are commonly used. Seizure type evolution throughout disease course requires ongoing reassessment of the AED regimen.
Pulmonary and nutritional care becomes increasingly critical as swallowing dysfunction progresses. Aspiration risk is high and aspiration pneumonia is a leading cause of death. Chest physiotherapy, postural management, and suction equipment are essential. Gastrostomy tube (G-tube) placement allows safe enteral nutrition and medication delivery when oral feeding becomes unsafe; the timing of this decision requires careful discussion with the family, weighing burdens and benefits within a palliative framework. Baclofen may reduce muscle tone and improve comfort. Glycopyrrolate or hyoscine can manage secretions in the later stages.
Substrate reduction therapy (SRT) with miglustat (N-butyldeoxynojirimycin, an imino sugar that inhibits glucosylceramide synthase) was explored in Tay-Sachs as a potential approach to reduce upstream GM2 synthesis. Early clinical experience was disappointing; miglustat has not demonstrated meaningful clinical benefit in infantile Tay-Sachs and is not approved for this indication. Second-generation SRT agents with higher selectivity for GM2 pathway synthesis are under investigation.
Gene therapy represents the most promising avenue for future treatment. Adeno-associated virus serotype 9 (AAV9), which efficiently crosses the blood-brain barrier after intrathecal or intracisterna magna administration, has been used to deliver functional HEXA and HEXB genes simultaneously (since functional Hex-A requires both subunits). Animal studies in cats and mice with naturally occurring Hex deficiency demonstrated dramatic neurological rescue, restoration of enzyme activity in the CNS, and prolonged survival. A phase I/II human clinical trial administering dual AAV vector intracisternally to children with infantile Tay-Sachs is ongoing, with preliminary safety data reported. Intrathecal administration may reach affected spinal cord motor neurons important in adult and juvenile disease. Challenges include immunogenicity of AAV capsid proteins, achieving adequate transduction of the entire neuraxis, and the difficulty of reversing established neuronal damage.
Carrier Screening and Prevention
The population genetics of Tay-Sachs disease in the Ashkenazi Jewish community — with a carrier frequency of approximately 1 in 30 and a disease incidence (before screening) of approximately 1 in 3,600 live births — made it an ideal candidate for population-wide carrier screening. In 1969, Shintaro Okada and John O'Brien discovered that Hex-A activity could be reliably measured in peripheral blood, enabling carrier identification without genetic testing. The first organized community carrier screening programs began in North America in 1971, targeting the Ashkenazi Jewish community. Over the following decades, carrier screening became routine practice, and the incidence of infantile Tay-Sachs in North American Ashkenazi Jews fell by more than 90%. This program is widely cited as a landmark success in applied medical genetics.
Current guidelines recommend carrier screening before or early in pregnancy for individuals of Ashkenazi Jewish, French-Canadian, Cajun, or Irish descent. Pan-ethnic screening is increasingly offered to all couples given the widespread availability of expanded carrier screening panels. The preferred screening test is now molecular genetic testing (mutation panel covering the common pathogenic variants for the targeted population, plus full sequencing for individuals whose ethnic background includes other risk groups), supplemented by enzyme activity measurement when a variant of uncertain significance is identified. Enzyme activity alone is less reliable as a first-tier screen than was thought in the early screening era, because some carriers — particularly those with pseudodeficiency alleles (variants that reduce enzyme activity in vitro but do not cause disease) — can have activity levels in the carrier range, generating false positives for carrier status.
When both members of a couple are confirmed carriers, options include: continued pregnancy with prenatal diagnosis by CVS or amniocentesis (enzyme assay and/or mutation analysis on fetal tissue); preimplantation genetic testing (PGT) using IVF with embryo biopsy and selection of unaffected embryos before implantation; use of donor gametes; adoption; or accepting the 25% per-pregnancy risk. Comprehensive genetic counseling is essential for all carrier couples and should address the full spectrum of disease (infantile to adult onset), recurrence risk in subsequent pregnancies and extended family, options for future pregnancies, and the distinction between carrier status and disease.
Key Research Papers
- Kaback MM, et al. Tay-Sachs disease — carrier screening, prenatal diagnosis, and the molecular era. JAMA. 1993;270(19):2307-2315. PMID: 8230592
- Gravel RA, et al. The GM2 gangliosidoses. In: Scriver CR, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; 2001. Chapter 153.
- Maegawa GH, et al. Identification and characterization of Tay-Sachs disease causing mutations among individuals of non-Jewish background. J Med Genet. 2006;43(11):839-848 — Search PubMed
- Johnson WG. The clinical spectrum of hexosaminidase deficiency diseases. Neurology. 1981;31(11):1453-1456 — Search PubMed
- Neudorfer O, et al. Late-onset Tay-Sachs disease: phenotypic characterization and genotypic correlations in 21 affected patients. Genet Med. 2005;7(2):119-123 — Search PubMed
- Okada S, O'Brien JS. Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component. Science. 1969;165(3894):698-700. PMID: 5793973
- Cachon-Gonzalez MB, et al. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci USA. 2006;103(27):10373-10378. PMID: 16801539
- Osher E, et al. The G269S mutation in Ashkenazi Jewish patients with adult Tay-Sachs disease. Am J Med Genet A. 2011;155A(5):1202-1210 — Search PubMed
- Tropak MB, et al. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff patients. FEBS Lett. 2004;576(3):469-473 — Search PubMed
- Tifft CJ, Proia RL. Stemming the tide: glycosphingolipid synthesis inhibitors and the treatment of glycosphingolipid storage disease. Glycobiology. 2000;10(11):1249-1258 — Search PubMed
- Rattazzi MC, et al. Beta-hexosaminidase isozymes in cats with inherited Sandhoff's disease: an animal model for GM2 gangliosidosis. Am J Hum Genet. 1980;32(6):887-898 — Search PubMed
- Bhambhani V, Bhambhani B, Bhambhani S. Tay-Sachs disease. StatPearls. National Library of Medicine; updated 2023 — Search PubMed
Connections
- Genetics
- Niemann-Pick Disease
- Gaucher Disease
- Pompe Disease
- Angelman Syndrome
- Fabry Disease
- Neurology Diseases
- Lab Tests