Insulinoma — Pancreatic Beta-Cell Tumor
Interactive Visualization Inside the Beta Cell — release insulin yourself Zoom inside a beta cell and watch glucose close the K-ATP channel, calcium flood in, and insulin granules pop out — then force it with a sulfonylurea or silence it with diazoxide. Launch →Table of Contents
- Overview
- The Rule of 90s
- Whipple's Triad and Clinical Presentation
- Pathophysiology
- Diagnosis — 72-Hour Supervised Fast
- Localization Imaging
- Treatment — Surgical Resection
- Medical Management and Malignant Insulinoma
- MEN1 Association
- Prognosis
- Key Research Papers
- Connections
- Featured Videos
Overview
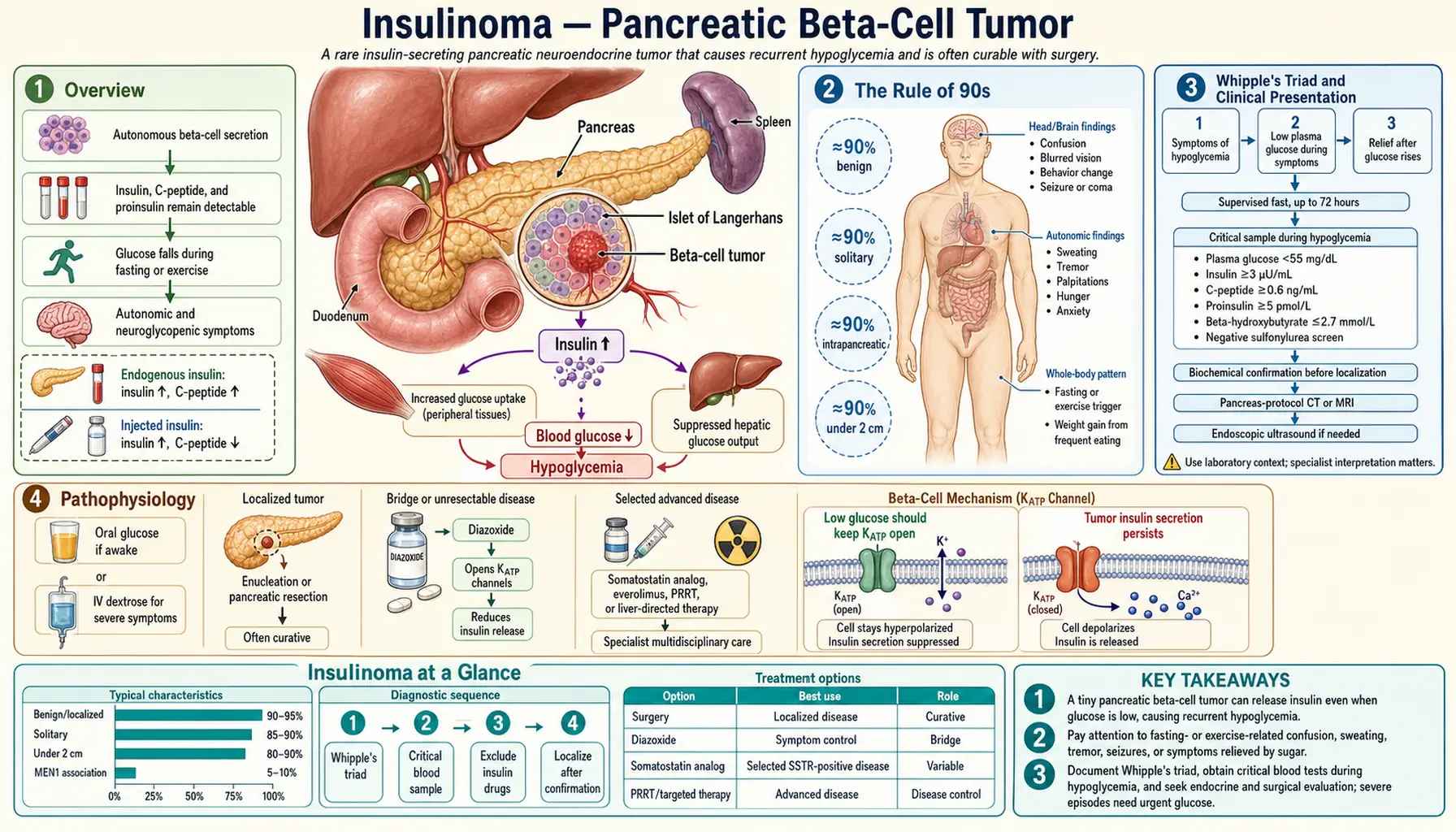
Insulinoma is the most common functional pancreatic neuroendocrine tumor (PNET), arising from the insulin-secreting beta cells of the pancreatic islets of Langerhans. Unlike normal beta cells, which tightly regulate insulin output in response to blood glucose levels, insulinoma cells secrete insulin autonomously — independent of whether glucose is high or low. The result is endogenous hyperinsulinemic hypoglycemia: chronically and dangerously low blood sugar driven by the tumor's unregulated insulin production.
Insulinoma is a rare condition, with an incidence of approximately 1 to 4 cases per million people per year. Despite its rarity, it is clinically important because it is a potentially curable cause of severe, recurrent hypoglycemia — and it is frequently misdiagnosed for months or years before the correct diagnosis is established. The peak age of presentation is between 40 and 60 years, with a slight female predominance. The tumor is distributed roughly equally throughout the pancreas — head, body, and tail — with no strong anatomic predilection.
Insulinomas belong to the broader category of pancreatic neuroendocrine tumors, which also includes gastrinomas (Zollinger-Ellison syndrome), glucagonomas, VIPomas, and somatostatinomas. Among all PNETs, insulinoma is unique in that it is the most commonly benign, the most commonly curable, and the most commonly misdiagnosed due to its neuropsychiatric presentation mimicking epilepsy, dementia, or psychiatric illness.
The Rule of 90s
The epidemiology of insulinoma is neatly summarized by the classic "Rule of 90s," a set of clinically memorable proportions that capture the key features of the disease:
- 90% are solitary — a single tumor is the rule; multiple tumors should immediately raise suspicion for Multiple Endocrine Neoplasia Type 1 (MEN1) or, less commonly, nesidioblastosis.
- 90% are benign — the vast majority are histologically and behaviorally benign adenomas, not carcinomas; malignancy is defined by local invasion or distant metastasis, not histologic appearance alone.
- 90% are less than 2 cm — this small size is the central challenge of management; standard cross-sectional imaging frequently misses these tumors, and specialized modalities are required.
- 90% are intrapancreatic — while ectopic insulinomas have been described (in duodenal wall, gastric wall, hilum of spleen, and liver), nearly all are within the pancreatic parenchyma itself.
The critical practical implication of these statistics is that functional biochemical confirmation must always precede localization imaging. Because 90% of these tumors are smaller than 2 cm, CT and MRI will miss a significant proportion on first pass. A negative imaging study in the face of biochemical proof of endogenous hyperinsulinism does not rule out insulinoma — it simply means the tumor has not yet been found. Proceeding to pancreatic surgery based on biochemistry alone (with intraoperative ultrasound to find the lesion) is established practice when imaging is negative.
Whipple's Triad and Clinical Presentation
The clinical diagnosis of insulinoma rests on Whipple's Triad, first described by the surgeon Allen Oldfather Whipple in 1935. The triad consists of three elements that must all be present:
- Symptoms of hypoglycemia during fasting or exercise — these fall into two categories. Neuroglycopenic symptoms result from glucose deprivation to the brain: confusion, cognitive impairment, slurred speech, bizarre behavior, personality change, amnesia, visual disturbances, seizures, and loss of consciousness. Autonomic (adrenergic) symptoms result from the counter-regulatory epinephrine surge: sweating, palpitations, tremor, anxiety, and pallor. In insulinoma, neuroglycopenic symptoms tend to predominate because hypoglycemia develops slowly (allowing partial adaptation), and the brain is the primary victim.
- Documented low plasma glucose at the time of symptoms — the Endocrine Society defines hypoglycemia as a plasma glucose below 55 mg/dL (3.0 mmol/L) at the time of symptoms; the historical threshold was below 45 mg/dL. A home glucometer reading is insufficient; laboratory plasma glucose confirmation is required.
- Relief of symptoms with glucose administration — symptoms must promptly resolve with oral or intravenous glucose administration. This differentiates true hypoglycemia from other causes of episodic neurological dysfunction.
The classic patient presentation is a middle-aged woman who has been experiencing episodic confusion, bizarre behavior, or apparent seizures for months to years, typically in the morning before breakfast or after exercise. A pattern that is almost pathognomonic: the patient has intuitively learned to eat very frequently to prevent episodes, and has gained significant weight as a direct consequence of the constant caloric intake needed to suppress the tumor's relentless insulin secretion. The weight gain in a patient complaining of "seizures" or "spells" is an important diagnostic clue.
The average time from symptom onset to correct diagnosis in published series is 1 to 2 years. Common misdiagnoses include epilepsy (neuroglycopenic seizures), transient ischemic attacks (focal neurological symptoms), psychiatric illness (personality change, bizarre behavior), dementia (cognitive impairment in older patients), and alcohol intoxication (disorientation, slurred speech). Every patient given an epilepsy diagnosis should have fasting glucose measured; prolonged EEG monitoring has been done unnecessarily in many insulinoma patients.
Pathophysiology
In normal physiology, pancreatic beta cells are exquisitely sensitive to blood glucose concentrations through the glucokinase enzyme, which acts as a "glucose sensor." As blood glucose falls during fasting, glucokinase activity decreases, ATP production in the beta cell drops, K-ATP channels open, the cell hyperpolarizes, calcium influx stops, and insulin secretion is suppressed. This elegant feedback mechanism prevents healthy beta cells from ever producing hypoglycemia in a non-diabetic person under normal circumstances.
In insulinoma, the tumor cells lose this regulated feedback. The glucokinase-driven glucose sensor is functionally uncoupled from secretory machinery, and insulin secretion continues autonomously even as plasma glucose falls into the hypoglycemic range. The molecular basis involves somatic mutations and epigenetic alterations in genes regulating beta-cell secretory thresholds, including YY1 mutations found in approximately 30% of sporadic insulinomas.
The metabolic consequences of autonomous insulin secretion are profound. Insulin drives glucose into muscle and fat cells, depletes hepatic glycogen stores (so glycogenolysis cannot rescue glucose levels), and suppresses gluconeogenesis (blocking the liver's backup glucose production). Simultaneously, insulin powerfully inhibits lipolysis and fatty acid beta-oxidation, which means the body cannot generate ketone bodies as an alternative brain fuel. This is the critical diagnostic clue: during hypoglycemia caused by insulinoma, ketones are LOW or absent. In starvation hypoglycemia, fasting hypoglycemia from other causes, or factitious hypoglycemia from exogenous insulin, this pattern differs — or ketones rise appropriately to protect the brain. The suppression of ketogenesis by endogenous insulin marks insulinoma as distinct from most other hypoglycemic disorders.
The counter-regulatory response — glucagon release from alpha cells, epinephrine from adrenal medulla, cortisol, and growth hormone — attempts to raise glucose but is overwhelmed by the tumor's continuous insulin output. With recurrent hypoglycemic episodes, counter-regulatory responses can become blunted (hypoglycemia unawareness), making episodes more dangerous and less symptomatic over time.
Diagnosis — 72-Hour Supervised Fast
The 72-hour supervised fast is the gold standard diagnostic test for insulinoma. It is performed in a monitored inpatient hospital setting where hypoglycemia can be safely induced and documented. The patient fasts completely from calories (water and non-caloric medications are permitted). Structured blood sampling occurs throughout the fast.
Sampling protocol: Every 6 hours during the fast: serum glucose, insulin, C-peptide, proinsulin, and beta-hydroxybutyrate (a ketone body). When plasma glucose falls below 60 mg/dL, sampling frequency increases to every 1 to 2 hours. The fast is terminated when: (1) plasma glucose falls below 45 mg/dL in the presence of symptoms, or (2) 72 hours have elapsed without hypoglycemia. At the endpoint, a critical sample set is collected simultaneously, then 1 mg glucagon is administered intravenously — a glucose rise of more than 25 mg/dL confirms adequate hepatic glycogen and inappropriate insulin action.
Positive diagnostic criteria for insulinoma (all should be present at the time of symptomatic hypoglycemia):
- Plasma glucose below 55 mg/dL (Endocrine Society threshold) with symptoms consistent with hypoglycemia.
- Serum insulin above 3 microU/mL — this appears low in isolation, but at the time of hypoglycemia, insulin should be fully suppressed toward zero; any detectable insulin is inappropriately elevated.
- Serum C-peptide above 0.6 ng/mL — C-peptide is cleaved from proinsulin during endogenous insulin synthesis. Elevated C-peptide at the time of hypoglycemia confirms that the insulin is being made inside the patient, not injected. Factitious hypoglycemia from exogenous insulin injection produces high insulin but suppressed C-peptide (since commercial insulin contains no C-peptide).
- Serum proinsulin above 5 pmol/L — insulinomas often secrete disproportionately large amounts of proinsulin (the uncleaved precursor), making elevated proinsulin a sensitive marker.
- Beta-hydroxybutyrate below 2.7 mmol/L — the absence of ketosis during hypoglycemia confirms that insulin is actively suppressing lipolysis and ketogenesis; this distinguishes insulinoma from starvation and most other fasting hypoglycemic disorders.
- Negative blood and urine screening for sulfonylureas and meglitinides — these oral hypoglycemic drugs can produce an identical biochemical picture (high insulin, high C-peptide, low ketones, low glucose) and must be excluded before diagnosing insulinoma.
Approximately 70% of insulinomas are diagnosed within 24 hours of fasting; 90% within 48 hours; and nearly all within 72 hours. A negative 72-hour fast effectively excludes insulinoma in nearly all cases. The combination of symptomatic hypoglycemia + inappropriately elevated insulin + elevated C-peptide + low ketones + negative drug screen constitutes a virtually certain biochemical diagnosis of endogenous hyperinsulinism — and insulinoma accounts for the vast majority of cases (with nesidioblastosis and post-bariatric hypoglycemia as rare alternatives).
Localization Imaging
Biochemical confirmation must always precede localization imaging. Attempting to find a small pancreatic tumor without first proving endogenous hyperinsulinism leads to incidental findings being misinterpreted and unnecessary surgery. Once the biochemical diagnosis is secure, a systematic imaging approach is used to find the lesion before surgery.
Endoscopic Ultrasound (EUS) is the single best modality for pancreatic insulinoma localization, with sensitivity of 90 to 95% in experienced hands. The endoscope is passed into the duodenum and stomach, placing the ultrasound transducer immediately adjacent to the pancreas with no intervening bowel gas. Insulinomas appear as hypoechoic, well-circumscribed nodules. EUS can detect lesions as small as 2 to 3 mm. Fine-needle aspiration can be performed for cytologic confirmation. The limitation is operator dependence — results vary substantially with endoscopist experience.
Triphasic contrast-enhanced CT (with dedicated arterial phase) achieves 75 to 80% sensitivity. Insulinomas are hypervascular tumors that enhance brightly during the arterial phase (approximately 35 seconds after contrast injection). Standard portal-venous phase CT misses many insulinomas. Thin-slice pancreatic protocol CT with arterial phase is essential. Small lesions in the pancreatic head overlying the duodenum are most often missed.
MRI of the pancreas with gadolinium and dedicated pancreatic sequences has sensitivity comparable to CT (75 to 85%). Insulinomas appear T2-hyperintense and show arterial enhancement. MRI avoids radiation and may better characterize equivocal CT findings. Diffusion-weighted imaging adds sensitivity for small lesions.
68Ga-DOTATATE PET/CT (somatostatin receptor scintigraphy) is the standard functional imaging for most PNETs, where somatostatin receptor expression is high. However, insulinomas are a notable exception: they tend to express lower levels of somatostatin receptors (particularly SSTR2) compared to gastrinomas and other PNETs. As a result, 68Ga-DOTATATE sensitivity for insulinoma is lower (approximately 50 to 70%), and a negative scan does not rule out insulinoma. GLP-1 receptor PET/CT using radiolabeled exendin is emerging as a more sensitive functional imaging approach specifically for insulinoma.
Arterial Stimulation with Hepatic Venous Sampling (ASVS) is an invasive but highly accurate regionalization technique used when cross-sectional imaging fails to locate the tumor. A calcium gluconate bolus (a potent insulin secretagogue acting on beta cells) is injected sequentially into the splenic artery, superior mesenteric artery, and gastroduodenal artery via angiographic catheter. Simultaneously, blood is sampled from the hepatic vein. A two-fold or greater step-up in hepatic venous insulin concentration following injection into a particular arterial territory identifies the vascular region harboring the tumor — localizing it to the tail (splenic artery territory), head (gastroduodenal artery territory), or body/uncinate (SMA territory). ASVS does not show the anatomic position of the tumor, but guides surgical exploration.
Intraoperative ultrasound is considered mandatory during surgical resection. Even when preoperative imaging has identified the lesion, the operating surgeon uses intraoperative ultrasound to confirm location, assess proximity to the main pancreatic duct (critical for deciding enucleation vs. resection), and ensure no additional tumors are missed. Combined with careful bimanual palpation of the pancreas, intraoperative ultrasound increases the tumor detection rate to above 97% in experienced centers.
Treatment — Surgical Resection
Surgery is the treatment of choice for insulinoma and offers the only potential cure. The surgical approach is tailored to tumor size, location, relationship to the main pancreatic duct, and the patient's overall fitness for surgery.
Enucleation is the preferred operation for small (less than 2 cm), superficially located, benign-appearing tumors that are not immediately adjacent to the main pancreatic duct. The tumor is shelled out from the surrounding pancreatic parenchyma with minimal loss of normal tissue. Intraoperative ultrasound is used to define the distance between the tumor and the main duct — less than 2 to 3 mm proximity is generally considered a contraindication to enucleation due to the risk of pancreatic fistula. Enucleation carries the lowest morbidity of any pancreatic operation and preserves maximal pancreatic function.
Distal pancreatectomy removes the body and tail of the pancreas and is the operation of choice for tumors in those locations that cannot be safely enucleated. It can often be performed laparoscopically, with lower complication rates than open surgery. Spleen-preserving distal pancreatectomy is preferred when technically feasible to avoid the long-term immune consequences of splenectomy.
Pancreaticoduodenectomy (Whipple procedure) is reserved for tumors in the pancreatic head that cannot be safely enucleated due to proximity to the main pancreatic duct or the pancreatic duct of Santorini. This is the highest-morbidity pancreatic operation, with significant rates of delayed gastric emptying, pancreatic fistula, and long-term exocrine and endocrine insufficiency. High-volume pancreatic surgery centers have substantially better outcomes than low-volume hospitals; referral to a center performing more than 20 Whipple procedures annually is recommended when this operation is anticipated.
Preoperative preparation: Patients should be maintained on diazoxide and/or frequent feeding regimens until surgery. IV dextrose infusion must be immediately available during anesthesia induction and throughout the operation. Hypoglycemia during surgery can be severe and life-threatening.
Postoperative glucose monitoring is essential in the immediate postoperative period. A characteristic pattern occurs: immediately after successful tumor resection, plasma glucose frequently rises sharply — sometimes into the diabetic range — due to the sudden loss of tumor insulin and the patient's heightened counter-regulatory state. This reactive hyperglycemia is transient and generally resolves within days to weeks as the normal beta-cell mass re-establishes regulated insulin secretion. Persistent postoperative hypoglycemia suggests incomplete resection or a missed second tumor.
Medical Management and Malignant Insulinoma
Medical management is used in three settings: (1) preoperative stabilization before planned surgery, (2) management of patients unfit for surgery, and (3) treatment of malignant insulinoma with metastatic disease not amenable to cure.
Diazoxide is the first-line medical therapy for symptomatic insulinoma. It acts by opening K-ATP channels on beta-cell membranes, hyperpolarizing the cell and directly inhibiting insulin secretion — the same mechanism that sulfonylureas activate in reverse. The typical dose is 150 to 400 mg per day in two to three divided oral doses. Response rates of 50 to 60% are seen. Major side effects include hirsutism (particularly troublesome for women), fluid retention and peripheral edema (managed with concurrent thiazide diuretics), and nausea. Diazoxide is not effective in malignant insulinoma due to altered receptor expression in tumor metastases.
Somatostatin analogs (octreotide, lanreotide) must be used with great caution in insulinoma and only after confirming somatostatin receptor expression, typically via somatostatin receptor scintigraphy. The rationale — inhibiting insulin secretion via somatostatin receptor activation — seems logical, but there is a critical hazard: somatostatin analogs also suppress glucagon secretion. Glucagon is a key counter-regulatory hormone that raises blood glucose during hypoglycemia. In insulinoma patients whose tumors express low somatostatin receptor levels (common), the net effect of octreotide can paradoxically worsen hypoglycemia by suppressing glucagon without adequately suppressing tumor insulin. Use only in receptor-positive tumors confirmed by imaging.
Frequent small meals with complex carbohydrates is the cornerstone behavioral intervention for all insulinoma patients while awaiting surgery or managing medically. Avoiding prolonged fasting (including overnight), eating every 2 to 3 hours, and ensuring a bedtime snack significantly reduce hypoglycemic episode frequency and severity. Uncooked cornstarch (a slowly digested polysaccharide) provides sustained glucose release and is particularly useful as a bedtime supplement.
Malignant insulinoma accounts for approximately 10% of cases and is defined by the presence of liver metastases (most common), lymph node involvement, or invasion of adjacent structures. Local recurrence after initial surgery can also represent malignant behavior. Management of malignant insulinoma requires a multidisciplinary approach:
- Everolimus — an oral mTOR (mammalian target of rapamycin) inhibitor; the RADIANT-3 trial demonstrated significantly improved progression-free survival (11 months vs. 4.6 months) in patients with advanced progressive PNETs including insulinoma. Glucose effects must be monitored: everolimus itself causes hyperglycemia by impairing insulin signaling, which can actually help control hypoglycemia in malignant insulinoma — but produces diabetic-range hyperglycemia if the tumor is subsequently controlled.
- Sunitinib — a VEGFR/PDGFR tyrosine kinase inhibitor; the Raymond et al. trial showed improved progression-free survival (11.4 months vs. 5.5 months) in advanced PNETs, with an overall survival benefit. An alternative to everolimus in malignant disease.
- 177Lu-DOTATATE (PRRT) — peptide receptor radionuclide therapy using a radiolabeled somatostatin analog to deliver targeted radiation to somatostatin receptor-expressing tumor cells; highly effective for receptor-positive PNETs, but limited by the variable somatostatin receptor expression in insulinoma. Patient selection requires receptor confirmation.
- Cytoreductive surgery and liver-directed therapy — surgical debulking of liver metastases, transarterial chemoembolization (TACE), and radiofrequency ablation can reduce tumor burden and improve hypoglycemia control even without cure.
- Streptozocin-based chemotherapy — streptozocin with 5-fluorouracil or doxorubicin has been used for malignant PNETs with modest response rates; largely supplanted by everolimus and sunitinib for first-line use.
MEN1 Association
Multiple Endocrine Neoplasia Type 1 (MEN1) is an autosomal dominant syndrome caused by loss-of-function mutations in the MEN1 gene on chromosome 11q13, which encodes the tumor suppressor protein menin. The classic clinical triad of MEN1 is: (1) primary hyperparathyroidism (parathyroid adenomas or hyperplasia, the most penetrant manifestation, occurring in more than 95% of patients by age 50), (2) pituitary adenomas (most commonly prolactinoma), and (3) pancreatic neuroendocrine tumors.
Approximately 5 to 10% of all insulinomas occur in the context of MEN1. The critical clinical clue that MEN1 may be present is the finding of multiple insulinomas — while sporadic insulinoma is almost always solitary, MEN1-associated PNETs are characteristically multiple. A patient with two or more insulinomas on imaging or at surgery should be considered to have MEN1 until proven otherwise and requires comprehensive workup: serum calcium (elevated in hyperparathyroidism), PTH (elevated), prolactin, IGF-1, and MEN1 genetic testing. First-degree relatives of confirmed MEN1 patients should also be screened.
MEN1-associated insulinomas present at a younger age (typically in the 20s to 30s) compared to sporadic insulinoma. They are more difficult to cure surgically because the entire pancreatic islet cell population carries the underlying genetic defect, and new tumors can emerge from any islet cell cluster over time. Surgical strategy is debated: some centers pursue aggressive resection to achieve euglycemia, while others accept medical management for smaller tumors given the near-certainty of recurrence after subtotal resection.
Gastrinomas (causing Zollinger-Ellison syndrome) are actually the most common pancreatic NET in MEN1 overall, but insulinoma is clinically significant because of the danger of hypoglycemia. In MEN1, the presence of one pancreatic NET should prompt screening for others: annual biochemical surveillance (gastrin, insulin, glucagon, VIP, pancreatic polypeptide) and periodic pancreatic imaging (EUS every 1 to 3 years) is recommended for known MEN1 carriers.
Prognosis
Benign solitary insulinoma carries an excellent prognosis. Surgical cure rates exceed 90% in experienced hands, and the vast majority of patients are completely cured with normalization of blood glucose after tumor resection. Operative mortality is less than 1% at high-volume pancreatic surgery centers. Long-term recurrence after apparently complete resection of sporadic benign insulinoma is uncommon, occurring in approximately 5 to 10% of patients over long-term follow-up (Service et al. 60-year Mayo Clinic series). Recurrence warrants MEN1 evaluation and repeat imaging.
Malignant insulinoma with metastatic disease has a substantially worse but variable prognosis. Five-year survival ranges from 25 to 65% depending on tumor burden, pace of progression, and availability of effective therapies. Insulinoma-related hypoglycemia can itself be life-threatening in metastatic disease, and controlling glucose is often the primary management priority. With modern targeted therapies (everolimus, sunitinib) and PRRT for receptor-positive tumors, survival has improved considerably compared to earlier decades when only cytotoxic chemotherapy was available.
MEN1-associated insulinoma requires indefinite surveillance regardless of initial surgical outcome. Even after what appears to be a curative resection, new PNETs can develop from remaining islet tissue carrying the MEN1 germline mutation. Annual biochemical surveillance and periodic pancreatic imaging are standard lifelong recommendations. The overall life expectancy of well-managed MEN1 patients has improved substantially with structured surveillance programs that identify and treat complications early.
Quality of life after curative surgery for benign insulinoma is generally excellent. Weight that was gained from constant eating to prevent hypoglycemia is typically lost postoperatively. Cognitive function, which may have been subtly impaired from recurrent hypoglycemic episodes, commonly improves after cure, though patients with prolonged diagnostic delay may have more residual cognitive effects from repeated severe hypoglycemia. Neuroglycopenic injury from severe or prolonged hypoglycemic episodes represents the most serious preventable complication of delayed diagnosis.
Key Research Papers
- Service FJ et al. Functioning insulinoma — incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 1991. — Search PubMed
- Hirshberg B et al. Forty-eight-hour fast: the diagnostic test for insulinoma. J Clin Endocrinol Metab 2000. — Search PubMed
- Placzkowski KA et al. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987–2007. J Clin Endocrinol Metab 2009. — Search PubMed
- Lam KY, Lo CY. Pancreatic endocrine tumour: a 22-year clinico-pathological experience with morphological, immunohistochemical observation and a review of the literature. Eur J Surg Oncol 1997. — Search PubMed
- Doherty GM et al. Resection of adrenal tumors from patients with multiple endocrine neoplasia type 1. Surgery 1991. — Search PubMed
- Vezzosi D et al. Insulinomas associated with multiple endocrine neoplasia type 1: a distinct group. Eur J Endocrinol 2015. — Search PubMed
- Okabayashi T et al. Diagnosis and management of insulinoma. World J Gastroenterol 2013. — Search PubMed
- Shin JJ, Gorden P, Libutti SK. Insulinoma: pathophysiology, localization and management. Future Oncol 2010. — Search PubMed
- Kann PH et al. Endoscopic ultrasound in patients with multiple endocrine neoplasia type 1: detection of pancreatic tumours. Eur J Endocrinol 2006. — Search PubMed
- Kulke MH et al. Everolimus in patients with advanced pancreatic neuroendocrine tumors. N Engl J Med 2011. — Search PubMed
- Raymond E et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011. PMID: 21306237
- Guettier JM et al. A prospective study comparing endoscopic ultrasound and computed tomography in the preoperative evaluation of patients with Zollinger-Ellison syndrome. Surgery 2008. — Search PubMed
PubMed Topic Searches:
- Insulinoma diagnosis and treatment
- Insulinoma 72-hour fasting test
- Pancreatic neuroendocrine tumor MEN1
- Endogenous hyperinsulinemic hypoglycemia
- Insulinoma endoscopic ultrasound localization
Connections
- Endocrinology
- Inside the Beta Cell: How Insulin Is Released — interactive animation
- Multiple Endocrine Neoplasia Type 1 (MEN1)
- Zollinger-Ellison Syndrome (Gastrinoma)
- Glucagonoma
- Carcinoid Tumor
- Diabetes
- Hypoglycemia Awareness and Prevention
- Pancreatic Neuroendocrine Tumors