Adrenal Incidentaloma
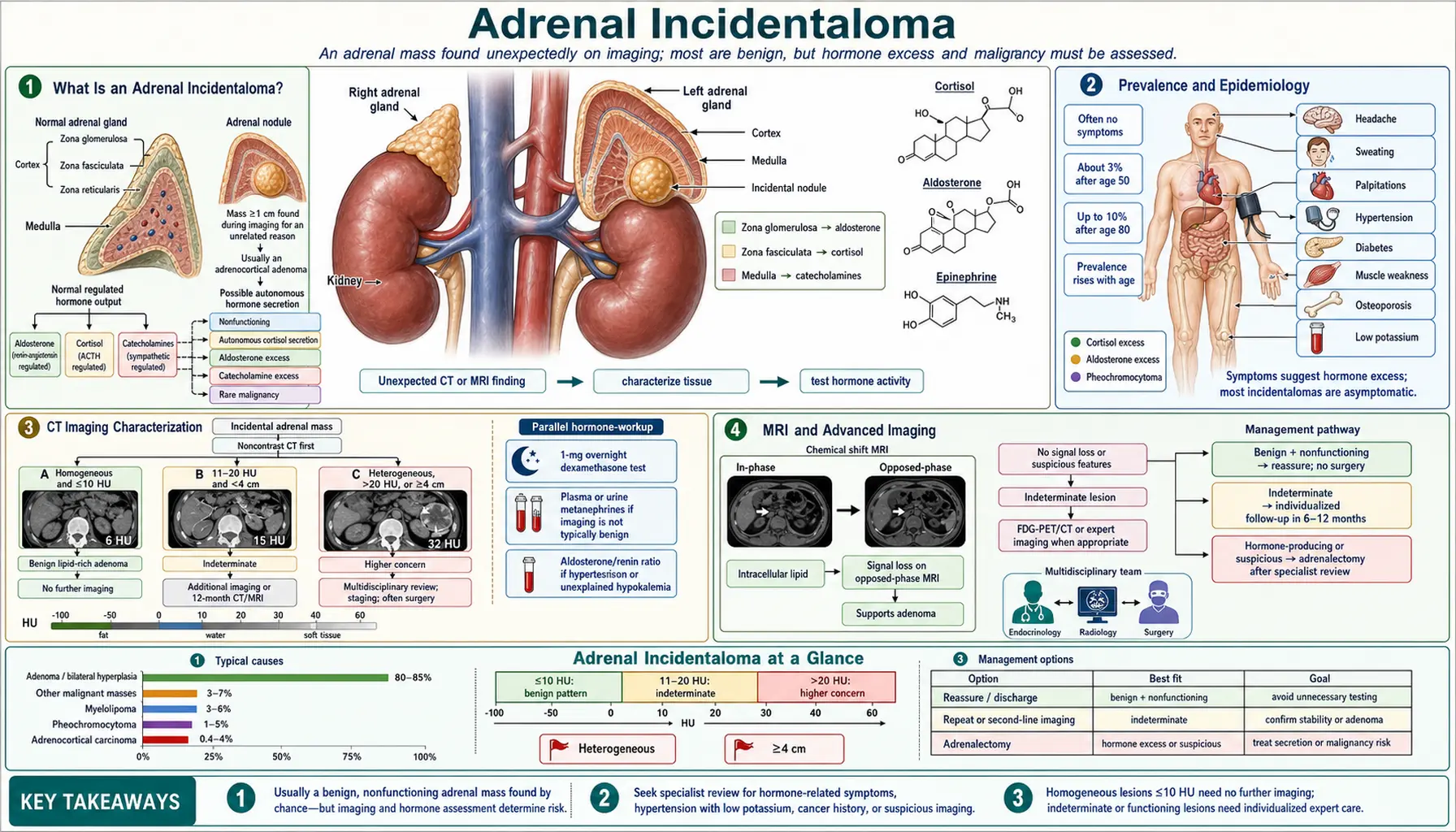
Table of Contents
- What Is an Adrenal Incidentaloma?
- Prevalence and Epidemiology
- CT Imaging Characterization
- MRI and Advanced Imaging
- Hormonal Evaluation
- Autonomous Cortisol Secretion
- Pheochromocytoma Workup
- Primary Aldosteronism Screening
- Adrenocortical Carcinoma
- Management Algorithm
- Key Research Papers
- Connections
- Featured Videos
What Is an Adrenal Incidentaloma?
An adrenal incidentaloma is an adrenal mass measuring 1 cm or larger discovered incidentally on imaging performed for a non-adrenal indication — such as a chest CT, abdominal CT for trauma, kidney stones, or pre-colonoscopy evaluation. The defining feature is that the mass was not the reason for the scan; it was simply found along the way.
The key clinical questions that guide every evaluation are: (1) Is the mass benign or malignant? (2) Is it hormonally active? The vast majority are benign non-functioning cortical adenomas, but the consequences of missing the dangerous exceptions are severe. Missing a pheochromocytoma before any surgical procedure can trigger a potentially fatal hypertensive crisis. Missing autonomous cortisol secretion means leaving a patient with untreated metabolic risk — hypertension, diabetes, bone loss — that could be addressed with surgery. Missing adrenocortical carcinoma (ACC) forfeits the only window for curative resection.
Every adrenal incidentaloma therefore triggers a structured two-part evaluation: imaging characterization to assess benignity vs. malignancy, and hormonal evaluation to assess function.
Prevalence and Epidemiology
Adrenal incidentalomas are common findings in an era of high-resolution cross-sectional imaging. CT-detected prevalence is approximately 1–2% in the overall adult population and rises to 3–7% in patients older than 70 years. Autopsy series report rates as high as 6%, confirming that many masses go undetected in life.
In the United States alone, an estimated 200,000 to 400,000 adrenal incidentalomas are discovered annually. Their distribution by diagnosis is well characterized:
- Benign non-functioning cortical adenoma: 70–80% of all incidentalomas.
- Autonomous cortisol secretion (ACS/mild autonomous CS): 10–20%; the most important functional finding.
- Pheochromocytoma: 3–7%; clinically the most dangerous if missed.
- Primary aldosteronism (Conn's adenoma): 1–2%, primarily in hypertensive patients.
- Adrenocortical carcinoma (ACC): 1–2%; rare but potentially curable only with early surgery.
- Metastases: approximately 2.5% in patients without known malignancy; 50–75% of adrenal masses in patients with a known primary cancer (lung, breast, melanoma, renal cell carcinoma are the most common primaries).
- Other benign lesions: myelolipoma (fat-containing, pathognomonic on CT), adrenal cyst, ganglioneuroma, hemangioma, neuroblastoma (pediatric).
Age and the presence of a known malignancy are the two strongest pre-test predictors of the underlying diagnosis.
CT Imaging Characterization
Unenhanced CT Hounsfield units (HU) are the first and most useful imaging tool for characterizing an adrenal incidentaloma:
- HU ≤10: lipid-rich adenoma. Sensitivity 71%, specificity 98%. More than 97% of masses with ≤10 HU are benign adenomas. No further imaging characterization is needed.
- HU 10–20: indeterminate — may be a lipid-poor adenoma or a malignancy; requires contrast washout or MRI chemical shift.
- HU >20 or heterogeneous: needs further characterization. Pheochromocytoma, ACC, and metastases all tend to be denser and more heterogeneous than adenomas.
For masses that do not meet unenhanced density criteria for adenoma, contrast-enhanced CT washout provides additional characterization:
- Absolute Percentage Washout (APW) = [(enhanced HU − delayed HU) / (enhanced HU − unenhanced HU)] × 100. APW ≥60% at 15 minutes = adenoma (sensitivity 88%, specificity 96%).
- Relative Percentage Washout (RPW) = [(enhanced HU − delayed HU) / enhanced HU] × 100. RPW ≥40% = adenoma (used when an unenhanced scan is unavailable).
Adenomas wash out contrast rapidly because they contain numerous functioning capillaries; malignancies retain contrast longer due to abnormal vascularity and neovasculogenesis.
Size matters: masses ≥4 cm raise concern for ACC. Masses ≥6 cm are strongly considered for surgery even when imaging appears benign, because the false-negative rate of imaging is not zero at this size. Features that increase malignancy suspicion regardless of size include irregular margins, calcification, heterogeneity, central necrosis, and local invasion.
MRI and Advanced Imaging
Chemical shift MRI exploits the fact that lipid-rich adenomas contain intracellular fat. On opposed-phase (OP) imaging, protons in fat and water cancel each other, causing signal loss relative to in-phase (IP) imaging. The signal intensity index (SII) is calculated as: SII = [(IP signal − OP signal) / IP signal] × 100. An SII >16.5% loss is diagnostic of a lipid-rich adenoma (sensitivity 81–87%, specificity 92–100%). Because MRI avoids ionizing radiation, it is preferred in younger patients and whenever repeated imaging is anticipated.
FDG-PET/CT measures glucose metabolism. An SUVmax ratio of incidentaloma to liver >1.45–3 indicates high metabolic activity consistent with malignancy (ACC or metastases). A low ratio supports benignity. FDG-PET/CT is particularly useful when: CT washout is indeterminate, the patient has a known malignancy, or the mass is heterogeneous and large. It is not used routinely for every incidentaloma.
18F-NaF PET can identify bone metastases when ACC or a metastatic primary is suspected. 68Ga-DOTATATE PET and MIBG scintigraphy are used for functional localization of pheochromocytoma and paraganglioma. Functional imaging is a confirmatory step after biochemical diagnosis — not a primary screening tool for all incidentalomas.
CT-guided biopsy is rarely useful for primary adrenal mass characterization (it cannot distinguish benign from malignant cortical tumors), but it may be appropriate when metastatic disease is suspected and the adrenal is the only accessible site for tissue diagnosis. Biopsy must be preceded by biochemical exclusion of pheochromocytoma to prevent hypertensive crisis.
Hormonal Evaluation
The 2023 European Society of Endocrinology/European Network for the Study of Adrenal Tumors (ESE/ENSAT) guidelines recommend hormonal evaluation for all adrenal incidentalomas regardless of size, with the exception of lesions with unambiguously benign imaging characteristics (simple cysts, myelolipomas with characteristic fat density). The rationale is that hormonal activity is not reliably predicted by size or imaging appearance.
The minimum workup consists of three tests:
- 1 mg overnight dexamethasone suppression test (DST) for autonomous cortisol secretion. Dexamethasone 1 mg is taken at 11 pm; serum cortisol is measured at 8 am the next morning. A cortisol >1.8 mcg/dL (50 nmol/L) indicates possible autonomous cortisol secretion (ACS). A cortisol >5 mcg/dL (138 nmol/L) indicates overt autonomous Cushing's syndrome.
- Plasma free metanephrines OR 24-hour urine fractionated metanephrines to exclude pheochromocytoma. This test must be done before any planned surgery or invasive procedure.
- Aldosterone-to-renin ratio (ARR) — only if the patient has hypertension, spontaneous hypokalemia, or diuretic-resistant hypokalemia. Routine ARR in normotensive patients without hypokalemia has a very low yield.
Additional tests added based on clinical suspicion: DHEAS (elevated in ACC or androgen-secreting tumor), 17-hydroxyprogesterone (17-OHP) if bilateral masses and concern for late-onset congenital adrenal hyperplasia, sex steroids (testosterone, estradiol, androstenedione) if clinical virilization or feminization is present.
Autonomous Cortisol Secretion
Autonomous cortisol secretion (ACS) — formerly called "subclinical Cushing's syndrome" — is the most clinically important functional finding in the workup of adrenal incidentalomas, and the label "subclinical" is increasingly recognized as a misnomer. ACS is not silent: it carries measurable metabolic and cardiovascular consequences even in the absence of the classic phenotypic features of Cushing's syndrome (moon face, buffalo hump, striae, proximal myopathy).
Prevalence in incidentaloma populations: post-1mg DST cortisol >1.8 mcg/dL is found in 20–50% of incidentalomas depending on the series. Using the higher threshold of >5 mcg/dL (overt CS), the rate is approximately 5–10%.
Established metabolic consequences of ACS:
- Hypertension: odds ratio 1.67 compared to non-functioning adenoma.
- Type 2 diabetes: odds ratio 2.1; impaired fasting glucose even higher.
- Osteoporosis/osteopenia: significantly elevated vertebral fracture rates.
- Dyslipidemia: elevated LDL, triglycerides; reduced HDL.
- Cardiovascular events and increased mortality in long-term follow-up studies.
Management of ACS: For patients with post-DST cortisol between 1.8 and 5.0 mcg/dL (mild ACS), first-line management is aggressive optimization of metabolic risk factors (antihypertensives, glucose control, bone protection). Adrenalectomy is considered for: younger patients (<40–50 years) with significant metabolic comorbidities attributable to ACS; patients with bilateral masses in whom the more active side can be identified; and patients with worsening comorbidities on conservative management. Adrenalectomy for ACS reduces blood pressure, improves glucose metabolism, and may reduce cardiovascular risk, though randomized trial data are limited. For overt ACS (>5 mcg/dL), adrenalectomy is generally recommended after excluding bilateral adrenal disease. Post-adrenalectomy glucocorticoid coverage is mandatory — contralateral adrenal suppression makes the patient temporarily adrenal-insufficient.
Pheochromocytoma Workup
Pheochromocytoma must be biochemically excluded before any surgical or invasive procedure involving an adrenal incidentaloma — this is a non-negotiable safety requirement. An unprepared pheochromocytoma can produce a life-threatening hypertensive crisis, malignant arrhythmia, or cardiovascular collapse when manipulated, biopsied, or exposed to contrast agents. The biochemical test is always first.
Biochemical tests:
- Plasma free metanephrines: sensitivity 99% for symptomatic pheochromocytoma; specificity 85–89%. Normetanephrine elevation is most common (norepinephrine-secreting tumors). False positives occur with certain antidepressants (tricyclics, SNRIs), acetaminophen, and adrenergic stress. Ideally drawn supine after 20–30 minutes of rest.
- 24-hour urine fractionated metanephrines and catecholamines: sensitivity 97%; slightly lower sensitivity than plasma for small tumors but higher specificity. Either test is acceptable as the initial screen.
Imaging characteristics of pheochromocytoma on CT/MRI: typically heterogeneous (due to hemorrhage and necrosis in larger tumors); unenhanced HU usually >20; contrast-enhanced CT shows avid early enhancement followed by delayed washout that does NOT meet the APW/RPW adenoma criteria. On MRI, pheochromocytoma classically shows very high T2 signal — the "light bulb sign" — due to high water content, though this finding is neither sensitive nor specific in all series. Large or malignant tumors may be cystic.
If pheochromocytoma is confirmed biochemically:
- Functional imaging for staging: 68Ga-DOTATATE PET (preferred for metastatic assessment) or 123I-MIBG scintigraphy.
- Genetic testing for hereditary syndromes (SDHx mutations, VHL, RET/MEN2, NF1) — up to 40% of pheochromocytomas have a germline mutation.
- Alpha-adrenergic blockade for 10–14 days preoperatively: phenoxybenzamine (non-selective, irreversible) or doxazosin/prazosin (selective alpha-1); high-sodium diet and liberal fluid intake to expand contracted intravascular volume.
- Beta-blockade only after alpha-blockade is established (to prevent unopposed alpha-stimulation and paradoxical hypertension).
- Laparoscopic adrenalectomy by an experienced surgeon is the preferred operative approach for most pheochromocytomas ≤6 cm.
Primary Aldosteronism Screening
Primary aldosteronism (PA) — also called Conn's syndrome — is not part of the routine workup for every incidentaloma. Screening is indicated when the patient has hypertension (particularly treatment-resistant hypertension), spontaneous hypokalemia (potassium <3.5 mEq/L without diuretics), or hypokalemia requiring >40 mEq/day supplementation despite removal of offending diuretics.
The screening test is the aldosterone-to-renin ratio (ARR). Cutoffs vary by assay and laboratory, but a commonly used threshold is: aldosterone ≥15 ng/dL with ARR ≥20–30 (when renin activity is in ng/mL/hr units) = positive screen. Many antihypertensive medications affect the ARR (ACE inhibitors and ARBs suppress aldosterone; beta-blockers suppress renin and can cause false positives). Ideally, patients are on medications that minimally affect the axis (calcium channel blockers, alpha-blockers, hydralazine) during testing.
Confirmatory testing options include the salt-loading test (24-hour urine aldosterone >12 mcg/24h after 3 days of high-sodium diet = PA confirmed) or the fludrocortisone suppression test (0.1 mg every 6 hours for 4 days; plasma aldosterone >6 ng/dL on day 4 = PA confirmed).
Adrenal vein sampling (AVS) is essential for subtype differentiation before any adrenalectomy. AVS distinguishes unilateral PA (aldosterone-producing adenoma — surgically curative via adrenalectomy) from bilateral adrenal hyperplasia (medical management with mineralocorticoid receptor antagonists: spironolactone or eplerenone). CT alone incorrectly classifies subtype in approximately 37% of patients with confirmed PA.
Successful curative adrenalectomy for unilateral PA resolves hypokalemia in essentially 100% of cases and achieves complete or partial blood pressure resolution in 50–70%, with significant reduction in cardiovascular risk.
Adrenocortical Carcinoma
Adrenocortical carcinoma (ACC) is rare — incidence 0.7–2 per million per year — but represents one of the most important diagnoses to exclude in the workup of an adrenal incidentaloma because the window for curative resection is narrow. Approximately 30–40% of ACCs present as incidentalomas; the remainder present with hormone excess (hypercortisolism, virilization) or mass effect.
Size remains the strongest imaging predictor of ACC:
- <4 cm: ACC risk approximately 2%
- 4–6 cm: ACC risk approximately 6%
- >6 cm: ACC risk approximately 25%
Additional imaging features raising concern: irregular margins, heterogeneous density, central necrosis, calcification, hemorrhage, local organ invasion, regional lymphadenopathy, venous invasion.
Staging (ENSAT system):
- Stage I: tumor ≤5 cm, confined to adrenal.
- Stage II: tumor >5 cm, confined to adrenal.
- Stage III: local infiltration or positive nodes.
- Stage IV: distant metastases.
Histological diagnosis uses the Weiss score (0–9 criteria including high nuclear grade, >5 mitoses per 50 HPF, atypical mitoses, <25% clear cells, diffuse architecture, necrosis, venous/sinusoidal/capsular invasion). A Weiss score ≥3 = malignant.
Treatment: Complete surgical resection with negative margins (R0) is the only potentially curative treatment. Open adrenalectomy — not laparoscopic — is mandated for suspected ACC to avoid capsule rupture and peritoneal seeding. Adjuvant mitotane is recommended for high-risk resected disease (stage III, high Ki-67 proliferation index, positive margins). For advanced/unresectable disease: mitotane plus EDP regimen (etoposide, doxorubicin, cisplatin). Median overall survival for metastatic ACC remains under 18 months.
5-year overall survival by stage: Stage I ~82%; Stage II ~61%; Stage III ~50%; Stage IV <13%.
Management Algorithm
The 2023 ESE/ENSAT guidelines streamlined the management of adrenal incidentalomas significantly compared to older protocols. The key change is that imaging follow-up is no longer required for clearly benign, non-functioning, small masses.
Clear benign adenoma (≤4 cm, HU ≤10 on unenhanced CT, non-functioning): No further imaging follow-up needed. This is a departure from the previous standard of 6- and 12-month repeat CT. Hormonal re-evaluation after 1 year is reasonable if new metabolic symptoms develop.
Indeterminate imaging, non-functioning, ≤4 cm: Repeat imaging at 6–12 months (CT or MRI). If stable in size and appearance, discharge from imaging surveillance. Hormonal re-evaluation at 1 year.
Any functioning adenoma (ACS, pheochromocytoma, primary aldosteronism, androgen excess): Treat per the relevant diagnosis. Decision for surgery vs. medical management depends on the specific functional diagnosis, severity, patient age, and comorbidities.
Indeterminate imaging, ≥4 cm, or malignancy concern: Multidisciplinary review (endocrinology + radiology + endocrine surgery). Options: adrenalectomy vs. interval imaging at 3–6 months vs. FDG-PET/CT. Biopsy is considered only after pheochromocytoma exclusion and when tissue diagnosis will change management (e.g., suspected metastasis from a known primary).
Indeterminate, ≥6 cm: Adrenalectomy strongly recommended even if imaging appears benign, because ACC cannot be reliably excluded at this size.
Bilateral incidentalomas: Additional considerations include late-onset congenital adrenal hyperplasia (measure morning 17-OHP), bilateral pheochromocytoma (rare; associated with MEN2, VHL, SDHx mutations), bilateral metastases, bilateral macronodular adrenal hyperplasia (BMAH) causing ACS, and bilateral PA (hyperplasia vs. bilateral adenomas — AVS critical).
Growth on surveillance: A size increase >1 cm per year on imaging is an indication for multidisciplinary reassessment and consideration of adrenalectomy, even for initially reassuring lesions.
Key Research Papers
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
- Search PubMed
Connections
- Endocrinology
- Cushing's Syndrome — the overt end of the cortisol spectrum that the 1 mg dexamethasone suppression test screens for.
- Pheochromocytoma — must be biochemically excluded before any biopsy or surgery on an adrenal mass.
- Primary Hyperaldosteronism — screened with the aldosterone-to-renin ratio when hypertension or hypokalemia is present.
- Adrenal Insufficiency — the expected post-adrenalectomy state when the contralateral gland has been suppressed.
- Congenital Adrenal Hyperplasia — the late-onset form to consider when incidentalomas are bilateral.