Polymyositis
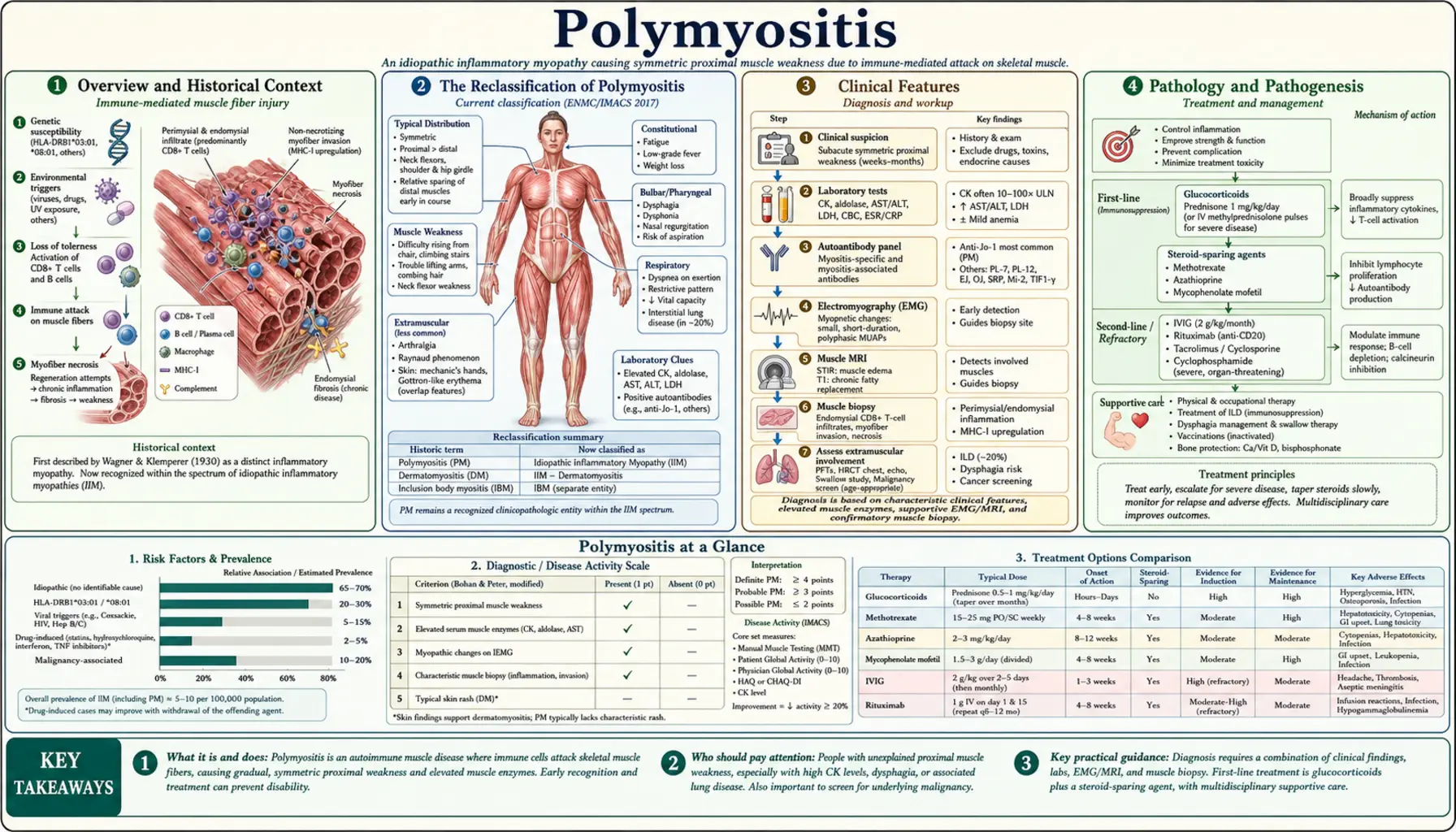
Polymyositis (PM) is an idiopathic inflammatory myopathy characterized by proximal muscle weakness, elevated muscle enzymes, and inflammatory infiltrates on biopsy. However, modern rheumatology has fundamentally reassessed whether "polymyositis" exists as a distinct entity. Most patients historically labeled with PM are now recognized to have more specific conditions — anti-synthetase syndrome, immune-mediated necrotizing myopathy (IMNM), or overlap myositis — each with distinct pathology, antibody profiles, and treatment responses. True PM, if it exists at all, is rare.
Table of Contents
- Overview and Historical Context
- The Reclassification of Polymyositis
- Clinical Features
- Pathology and Pathogenesis
- Differential Diagnosis
- Myositis Antibodies in PM
- Diagnosis and Workup
- Treatment and Response
- Prognosis and Complications
- Key Research Papers
Overview and Historical Context
Polymyositis was formally described in the medical literature in the late 19th and early 20th centuries as an inflammatory disorder of skeletal muscle without the skin findings seen in dermatomyositis. Ernst Wagner first used the term "polymyositis" in 1863. For much of the 20th century, PM was grouped with dermatomyositis (DM) and inclusion body myositis (IBM) under the umbrella of idiopathic inflammatory myopathies (IIMs).
The classic diagnostic criteria proposed by Bohan and Peter in 1975 — proximal muscle weakness, elevated CK, myopathic EMG, abnormal muscle biopsy, and absence of skin rash — were used for decades to define PM. Under these broad criteria, PM became a relatively common rheumatologic diagnosis. However, as myositis-specific antibody testing and more refined biopsy techniques became available, it became clear that many patients diagnosed with "PM" actually had distinct pathologic entities with different underlying mechanisms, clinical trajectories, and treatment responses.
The 2017 EULAR/ACR classification criteria represented a major step forward, incorporating antibody data and distinguishing IIM subtypes more precisely. Contemporary rheumatologists approach a suspected PM diagnosis with considerable skepticism, using it only as a provisional category while pursuing thorough antibody testing and biopsy interpretation to identify the true underlying condition.
The Reclassification of Polymyositis
The concept of "polymyositis" as a distinct disease is now seriously questioned. A landmark analysis by Dalakas and others demonstrated that most patients historically diagnosed with PM, when re-evaluated with modern antibody testing and refined biopsy criteria, could be reclassified into one of three better-defined entities:
- Anti-synthetase syndrome — Patients with antibodies to aminoacyl-tRNA synthetases (anti-Jo-1, anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS, anti-Zo, anti-Ha). These patients have myositis, interstitial lung disease, inflammatory arthritis, mechanic's hands, Raynaud's phenomenon, and fever. The lung disease is often the most serious complication. This entity accounts for a significant proportion of former "PM" cases.
- Immune-mediated necrotizing myopathy (IMNM) — Characterized by anti-SRP or anti-HMGCR antibodies, severe necrosis on muscle biopsy with minimal inflammatory infiltrate, and often statin association (anti-HMGCR). These patients have severe proximal weakness, very high CK levels, and require aggressive immunosuppression. The biopsy looks dramatically different from "classic PM."
- Overlap myositis — Myositis occurring in the setting of another connective tissue disease (CTD) such as systemic sclerosis, systemic lupus erythematosus, or Sjögren's syndrome. Anti-PM-Scl, anti-Ku, and anti-U1-RNP antibodies are associated with overlap myositis.
What remains after excluding these three categories — "true PM" — is characterized by endomysial CD8+ T cell infiltration directly invading non-necrotic muscle fibers, with MHC class I upregulation. This pure T cell-mediated disease is now considered rare, and some experts question whether it ever constitutes more than a small minority of IIM cases.
Clinical Features
When PM is suspected, the clinical presentation typically includes the following features:
Muscle Involvement
- Proximal muscle weakness — The hallmark: difficulty rising from a chair, climbing stairs, lifting arms above the head, combing hair. The shoulder girdle and hip girdle muscles are preferentially affected.
- Symmetric distribution — Unlike inclusion body myositis, weakness is generally symmetric.
- Muscle pain and tenderness — Present in some patients but not universal; significant pain should prompt reconsideration of other diagnoses.
- Distal sparing (early) — Hand grip and foot dorsiflexion are typically preserved until late in disease, distinguishing PM from IBM.
- Dysphagia — Pharyngeal and upper esophageal muscle involvement can occur, leading to dysphagia and aspiration risk.
Absence of Skin Findings
By definition, PM does not have the characteristic skin findings of dermatomyositis. The absence of heliotrope rash (periorbital violaceous discoloration), Gottron's papules (papules over the knuckles), V-sign (anterior chest/neck rash), shawl sign, or mechanic's hands is required. The presence of any of these features should redirect the diagnosis to DM, anti-synthetase syndrome, or overlap myositis.
Systemic Manifestations
- Interstitial lung disease (ILD) — Can occur in PM, but its presence strongly suggests anti-synthetase syndrome and should trigger antibody testing for anti-Jo-1 and other synthetase antibodies.
- Cardiac involvement — Myocarditis, conduction abnormalities, and cardiomyopathy can occur in inflammatory myopathies, including PM.
- Arthritis — Inflammatory polyarthritis can accompany PM, particularly in anti-synthetase syndrome.
- Raynaud's phenomenon — Suggests an overlap syndrome or anti-synthetase syndrome.
Pathology and Pathogenesis
The histopathologic features of PM on muscle biopsy are distinct from those of dermatomyositis, IBM, and IMNM, and correct interpretation is essential for accurate diagnosis.
Classic PM Biopsy Findings
- Endomysial inflammatory infiltrate — CD8+ cytotoxic T lymphocytes and macrophages in the endomysium (connective tissue between individual muscle fibers), invading and surrounding non-necrotic muscle fibers. This is the pathognomonic feature.
- MHC class I upregulation — Upregulation of MHC class I antigen on the sarcolemma (muscle fiber surface) of non-necrotic fibers, making them targets for CD8+ T cells. In normal muscle, MHC class I is not expressed on myofibers.
- Absence of perifascicular atrophy — Perifascicular atrophy (selective atrophy of muscle fibers at the periphery of fascicles) is a cardinal feature of dermatomyositis and should NOT be present in PM.
- Absence of rimmed vacuoles — Rimmed vacuoles (vacuoles lined with basophilic granular material containing p62, TDP-43, and other inclusions) are the hallmark of IBM and must be absent in PM.
- No extensive necrosis without inflammation — Extensive necrosis with minimal inflammation characterizes IMNM, not PM.
Pathogenesis
In true PM, the pathogenesis is T cell-mediated. CD8+ cytotoxic T cells recognize an antigen presented on MHC class I molecules on the surface of muscle fibers. The exact autoantigen(s) remain unknown, but the process involves clonal expansion of antigen-specific T cells that directly invade and destroy muscle fibers. This differs fundamentally from dermatomyositis, which is a humorally-mediated, complement-dependent microangiopathy primarily affecting the capillaries within muscle fascicles, driven by CD4+ T cells and B cells.
In IMNM, the pathogenesis involves autoantibodies (anti-SRP or anti-HMGCR) that directly or indirectly cause muscle fiber necrosis. In anti-synthetase syndrome, the mechanisms are multifactorial, involving both cellular and humoral responses targeting aminoacyl-tRNA synthetase enzymes.
Differential Diagnosis
The differential diagnosis of suspected PM is one of the most important clinical exercises in inflammatory myopathy. Getting this right determines treatment choice, prognosis, and cancer screening needs.
1. Immune-Mediated Necrotizing Myopathy (IMNM)
IMNM presents with severe proximal weakness and markedly elevated CK (often 5,000–50,000 IU/L). Key distinguishing features:
- Anti-SRP or anti-HMGCR antibodies (present in ~60–70% of IMNM cases)
- Biopsy shows extensive necrosis and regeneration with minimal inflammatory infiltrate — nearly the opposite of PM
- Often statin-associated (anti-HMGCR), but can occur without statin exposure
- Requires more aggressive immunosuppression than PM; rituximab or IVIG often needed
- Higher cancer risk, particularly with anti-SRP
2. Anti-Synthetase Syndrome
Often misdiagnosed as PM because muscle weakness is prominent. Key distinguishing features:
- Antibodies to aminoacyl-tRNA synthetases: anti-Jo-1 is most common (~20% of IIM), anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS
- Interstitial lung disease (ILD) — present in 60–80%; can be severe and life-threatening
- Mechanic's hands — hyperkeratotic, cracked skin on the lateral and palmar aspects of the fingers
- Inflammatory arthritis — often polyarticular, symmetric, non-erosive
- Raynaud's phenomenon
- Fever at disease onset
3. Inclusion Body Myositis (IBM)
The most common inflammatory myopathy in patients over 50. Key distinguishing features:
- Distal weakness prominent, especially finger flexors and foot extensors — the opposite pattern from PM
- Weakness is often asymmetric
- Quadriceps weakness and wasting prominent
- Dysphagia common and early
- Affects older men more than women (unlike PM/DM)
- Biopsy shows rimmed vacuoles, congophilic inclusions, and endomysial inflammation
- Poor response to immunosuppressive therapy — a key clinical clue; PM should improve with steroids
4. Dermatomyositis (DM)
Distinguished by characteristic skin findings (heliotrope rash, Gottron's papules, V-sign, shawl sign) and perifascicular atrophy on biopsy. The inflammatory infiltrate is perivascular and perimysial (not endomysial), driven by complement and type I interferon. Higher cancer association than PM (especially anti-NXP2, anti-TIF1γ antibodies).
5. Muscular Dystrophies
Limb-girdle muscular dystrophies (especially LGMD2B/dysferlinopathy and LGMD2A/calpainopathy) can mimic PM clinically. Key distinguishing features:
- Positive family history (though recessive forms may be sporadic)
- Earlier onset, slower progression
- Genetic testing for mutations in DYSF, CAPN3, SGCA/B/C/D, and other dystrophy genes
- Biopsy: dystrophic pattern without primary inflammatory infiltrate (though secondary inflammation can occur in dysferlinopathy)
- Poor response to immunosuppression
6. Metabolic Myopathies
Glycogen storage diseases (Pompe disease, McArdle disease), lipid storage myopathies (carnitine deficiency), and mitochondrial myopathies can present with muscle weakness and elevated CK. Exertional component, exercise intolerance, cramps, and myoglobinuria with exercise suggest metabolic causes. Enzyme assays and genetic testing establish diagnosis.
7. Drug-Induced Myopathy
Multiple drugs can cause inflammatory or necrotizing myopathy:
- Statins — Cause a spectrum from asymptomatic CK elevation to IMNM (anti-HMGCR). Stopping the statin resolves most statin-related myopathy, but IMNM can persist or worsen after statin discontinuation — a critical distinguishing point.
- Hydroxychloroquine — Vacuolar myopathy; biopsy shows lysosomal inclusions
- Colchicine — Proximal weakness, vacuolar myopathy on biopsy, particularly in renal impairment
- Checkpoint inhibitors — Immune-related myositis; increasingly recognized with pembrolizumab, nivolumab, ipilimumab
- Alcohol — Acute alcoholic myopathy, subacute alcoholic myopathy
Myositis Antibodies in PM
Myositis-specific antibodies (MSAs) and myositis-associated antibodies (MAAs) are critical for classifying inflammatory myopathies and have largely supplanted the old Bohan-Peter criteria. The absence of known MSAs in a patient with suspected PM is a diagnostic challenge; their presence almost always reclassifies the patient.
Myositis-Specific Antibodies (MSAs)
- Anti-Jo-1 (anti-histidyl-tRNA synthetase) — Most common antisynthetase antibody (~20% of IIM); defines anti-synthetase syndrome; associated with ILD, mechanic's hands, arthritis
- Anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS — Other synthetase antibodies; similar clinical syndrome to anti-Jo-1 but ILD may predominate more and myositis may be less prominent
- Anti-SRP (signal recognition particle) — IMNM; severe rapidly progressive myopathy, very high CK, cardiac involvement, poor treatment response
- Anti-HMGCR (3-hydroxy-3-methylglutaryl-CoA reductase) — IMNM; statin-associated; responds better to immunotherapy than anti-SRP IMNM
- Anti-Mi-2 — Strongly associated with classic dermatomyositis; V-sign and shawl sign prominent; good prognosis
- Anti-NXP-2, anti-TIF1γ — DM; high cancer association
- Anti-MDA5 (melanoma differentiation-associated gene 5) — Clinically amyopathic DM; rapidly progressive ILD; skin ulcers; high mortality risk
- Anti-SAE (small ubiquitin-like modifier activating enzyme) — DM; dysphagia prominent
Myositis-Associated Antibodies (MAAs)
- Anti-PM-Scl — Overlap myositis with systemic sclerosis; PM-Scl75 and PM-Scl100
- Anti-Ku — Overlap myositis with systemic sclerosis or SLE
- Anti-U1-RNP — Mixed connective tissue disease; myositis, pulmonary hypertension, Raynaud's
- Anti-Ro52 (TRIM21) — Associated with multiple IIM subtypes; higher ILD risk; often co-occurs with antisynthetase antibodies
Patients with clinical features of PM but without any identifiable MSA or MAA — so-called "seronegative PM" — represent the most uncertain diagnostic category. Careful repeat biopsy interpretation by a neuromuscular pathologist and close longitudinal follow-up are essential. Some will ultimately be reclassified as IBM, dystrophy, or another entity.
Diagnosis and Workup
The diagnosis of PM requires excluding the more common conditions that mimic it. The workup should proceed systematically:
Laboratory Testing
- Creatine kinase (CK) — Elevated in active PM, often 5–50 times upper limit of normal; very high levels (>10,000 IU/L) suggest IMNM
- Aldolase, LDH, AST, ALT — Additional muscle enzyme markers; useful when CK is only mildly elevated
- Comprehensive myositis antibody panel — Anti-Jo-1, anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS, anti-SRP, anti-HMGCR, anti-Mi-2, anti-NXP-2, anti-TIF1γ, anti-MDA5, anti-SAE, anti-PM-Scl, anti-Ku, anti-Ro52, anti-U1-RNP
- ANA (antinuclear antibody) — Screening for CTD overlap
- CBC, CMP, ESR, CRP — Baseline inflammatory markers and organ function
- Troponin, BNP — Cardiac involvement screening
- Aldolase and urinalysis — Myoglobinuria indicates severe ongoing muscle destruction
Electromyography (EMG)
EMG in PM shows a myopathic pattern: short-duration, small-amplitude, polyphasic motor unit action potentials with early recruitment. Spontaneous activity (fibrillations, positive sharp waves) reflects active denervation-like changes from muscle fiber necrosis. EMG is useful to confirm a myopathic process but does not distinguish PM from other IIMs.
MRI of Muscles
Muscle MRI (STIR sequences) demonstrates edema-like changes in affected muscles, helping to identify areas of active inflammation for biopsy targeting. MRI can also detect fatty infiltration reflecting chronic damage and guide follow-up assessment.
Muscle Biopsy
Muscle biopsy is essential and should be performed by an experienced neuromuscular pathologist. Key steps:
- Select a muscle with moderate (not end-stage) weakness — avoid the EMG-studied side to prevent sampling artifact
- MRI guidance improves diagnostic yield by targeting areas of active inflammation
- Immunohistochemistry for CD4, CD8, CD20, MHC class I/II, complement (C5b-9/MAC), and p62/TDP-43 is now standard
- Electron microscopy may be needed to identify IBM inclusions or mitochondrial abnormalities
Chest Imaging and PFTs
HRCT of the chest is recommended at baseline to screen for ILD, which has treatment and prognostic implications. Pulmonary function tests (spirometry, DLCO) quantify ILD severity if present.
Cancer Screening
The cancer association in PM is less pronounced than in DM but still present. Age-appropriate cancer screening (colonoscopy, mammography, PSA, Pap) is recommended at diagnosis. If anti-TIF1γ, anti-NXP-2, or anti-SRP antibodies are present, more intensive cancer screening is warranted.
The 2017 EULAR/ACR Classification Criteria
The 2017 EULAR/ACR criteria assign probability scores to individual features (onset age, muscle weakness pattern, skin findings, other organ involvement, CK level, biopsy features, antibodies) to classify a patient as having an IIM and determine the most probable subtype. This replaced the older Bohan-Peter criteria and better separates PM from DM, IBM, IMNM, and anti-synthetase syndrome.
Treatment and Response
Treatment of PM (and reclassified entities previously labeled PM) involves immunosuppressive therapy. The specific regimen depends on disease severity, antibody subtype, and organ involvement.
First-Line: Corticosteroids
High-dose prednisone (1 mg/kg/day, up to 60–80 mg/day) remains the first-line treatment. Most patients respond within 4–8 weeks with improving muscle strength and declining CK. In severe disease with dysphagia, respiratory involvement, or cardiac involvement, IV methylprednisolone pulse therapy (1 g/day for 3 days) may be used. Steroids are slowly tapered over 9–12 months as clinical improvement is maintained. Approximately 20–30% of patients achieve sustained remission with steroids alone, but most require steroid-sparing agents.
Steroid-Sparing Agents
- Methotrexate (MTX) — 15–25 mg/week; first-line steroid-sparing agent; effective for muscle disease; use with caution if ILD is present (risk of MTX-induced pneumonitis)
- Azathioprine (AZA) — 2–3 mg/kg/day; similar efficacy to MTX; preferred when ILD is present; requires TPMT testing before initiation
- Mycophenolate mofetil (MMF) — 2–3 g/day; particularly useful in ILD-dominant disease and overlap myositis
- Hydroxychloroquine — Used in overlap myositis with SLE/Sjögren's features; not effective as monotherapy for myositis
- Cyclosporine — Second-line; particularly used in anti-synthetase syndrome with ILD
- Tacrolimus — Alternative to cyclosporine; used in refractory ILD
Intravenous Immunoglobulin (IVIG)
IVIG (2 g/kg per cycle) is effective as rescue therapy for refractory PM and DM. A randomized controlled trial demonstrated benefit in DM, and IVIG is widely used in PM refractory to conventional immunosuppression. It is also the preferred treatment during pregnancy and in patients with severe dysphagia or respiratory compromise where rapid improvement is needed.
Biologic Agents
- Rituximab — Anti-CD20; the RIM trial showed benefit in refractory PM/DM; particularly used in antibody-positive cases (anti-Jo-1, anti-Mi-2, anti-PM-Scl); now considered for IMNM refractory to conventional treatment
- Abatacept — Anti-CTLA-4 fusion protein; explored in PM/DM with some positive results
- JAK inhibitors (tofacitinib, ruxolitinib) — Emerging data in refractory IIM, particularly DM with ILD; type I interferon pathway involvement makes JAK inhibition a rational target
Special Considerations by Subtype
- Anti-synthetase syndrome with ILD — MMF or AZA preferred over MTX; consider cyclosporine or tacrolimus for progressive ILD; rituximab for refractory cases
- IMNM (anti-SRP/anti-HMGCR) — Requires more aggressive initial treatment than PM; combination of high-dose steroids + AZA or MTX + IVIG; rituximab early in anti-SRP IMNM
- Overlap myositis — Treatment directed at both the myositis component and the underlying CTD
Non-Pharmacologic Treatment
Physical and occupational therapy are important adjuncts. Supervised exercise therapy — once thought potentially harmful in inflammatory myopathy — has been shown to be safe and beneficial in stable PM/DM, improving functional capacity without exacerbating inflammation. Dysphagia management by a speech-language pathologist is important for patients with pharyngeal involvement.
Prognosis and Complications
The prognosis of PM depends heavily on the correct underlying diagnosis and on the extent of extra-muscular involvement.
Disease Course
True PM (endomysial CD8+ T cell-mediated) generally has a better prognosis than IMNM or anti-synthetase syndrome, but relapses are common when steroids are tapered. A monophasic course occurs in roughly 20% of patients; most experience a relapsing-remitting or chronic persistent course. IBM has the worst prognosis among the IIMs in terms of functional disability — it progresses inexorably despite treatment.
Pulmonary Complications
ILD is the most important determinant of long-term morbidity and mortality in inflammatory myopathies. Rapidly progressive ILD (particularly in anti-MDA5 DM or anti-SRP IMNM) can be fatal within months. In anti-synthetase syndrome, ILD is chronic and may progress independently of myositis activity. Pulmonary hypertension can develop as a late complication. Aspiration pneumonia from pharyngeal weakness is a significant cause of acute respiratory deterioration.
Cancer Association
Inflammatory myopathies, particularly DM, are associated with increased cancer risk. In PM, the cancer association is more modest but still significant, with a standardized incidence ratio (SIR) estimated at 1.5–2.0 in large cohort studies. Lung, ovarian, colorectal, and NHL cancers are most commonly reported. The cancer risk is highest in the first 3 years after IIM diagnosis. Anti-TIF1γ antibodies confer the highest cancer risk in DM.
Cardiovascular Complications
Myocarditis occurs in up to 9% of IIM patients and may be clinically silent. Conduction abnormalities, arrhythmias, and dilated cardiomyopathy are recognized complications. Cardiovascular mortality is increased in IIM patients partly due to myocarditis and partly due to accelerated atherosclerosis from systemic inflammation and chronic corticosteroid use.
Mortality
Five-year survival in PM/DM has improved with modern immunosuppression but remains impaired compared to the general population. Causes of death include ILD, cancer, infection (particularly Pneumocystis jirovecii pneumonia — PCP prophylaxis with trimethoprim-sulfamethoxazole should be considered in patients on high-dose steroids), and cardiovascular disease. Older age at diagnosis, ILD, dysphagia, and cancer are the main adverse prognostic factors.
Key Research Papers
- Lundberg IE, Tjärnlund A, Bottai M, et al. "2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies." Ann Rheum Dis. 2017 — Search PubMed
- Hoogendijk JE, Amato AA, Lecky BR, et al. "119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies." Neuromuscul Disord. 2004 — Search PubMed
- Dalakas MC. "Inflammatory muscle diseases." N Engl J Med. 2015 — Search PubMed
- Fiorentino D, Chung L, Zwerner J, et al. "Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ." Arthritis Rheum. 2013 — Search PubMed
- Hallowell RW, Danoff SK. "Interstitial lung disease associated with the idiopathic inflammatory myopathies and the antisynthetase syndrome." Curr Opin Rheumatol. 2014 — Search PubMed
- Rider LG, Miller FW. "Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies." JAMA. 2011 — Search PubMed
- Danieli MG, Pettinari L, Moretti R, et al. "Intravenous immunoglobulin in polymyositis and dermatomyositis." Autoimmunity. 2014 — Search PubMed
- Aggarwal R, Oddis CV. "Therapeutic advances in myositis." Curr Opin Rheumatol. 2012 — Search PubMed
- Mammen AL. "Necrotizing autoimmune myopathy: beyond statins." Curr Opin Rheumatol. 2014 — Search PubMed
- Marie I. "Morbidity and mortality in adult polymyositis and dermatomyositis." Curr Rheumatol Rep. 2012 — Search PubMed
- Lundberg IE, de Visser M, Werth VP. "Idiopathic inflammatory myopathies." Nat Rev Dis Primers. 2021 — Search PubMed
- Allenbach Y, Keraen J, Bouvier AM, et al. "High risk of cancer in autoimmune necrotizing myopathies." Neurology. 2016 — Search PubMed
PubMed Topic Searches
- Polymyositis diagnosis and treatment — PubMed
- Idiopathic inflammatory myopathy classification — PubMed
- Anti-synthetase syndrome and ILD — PubMed
- Immune-mediated necrotizing myopathy — PubMed
Connections
- Rheumatology
- Dermatomyositis
- Inclusion Body Myositis
- Lupus
- Rheumatoid Arthritis
- Interstitial Lung Disease
- Anti-Synthetase Syndrome — the antibody-defined myositis that accounts for many historical polymyositis diagnoses.