IgA Deficiency (Selective IgA Deficiency)
Overview
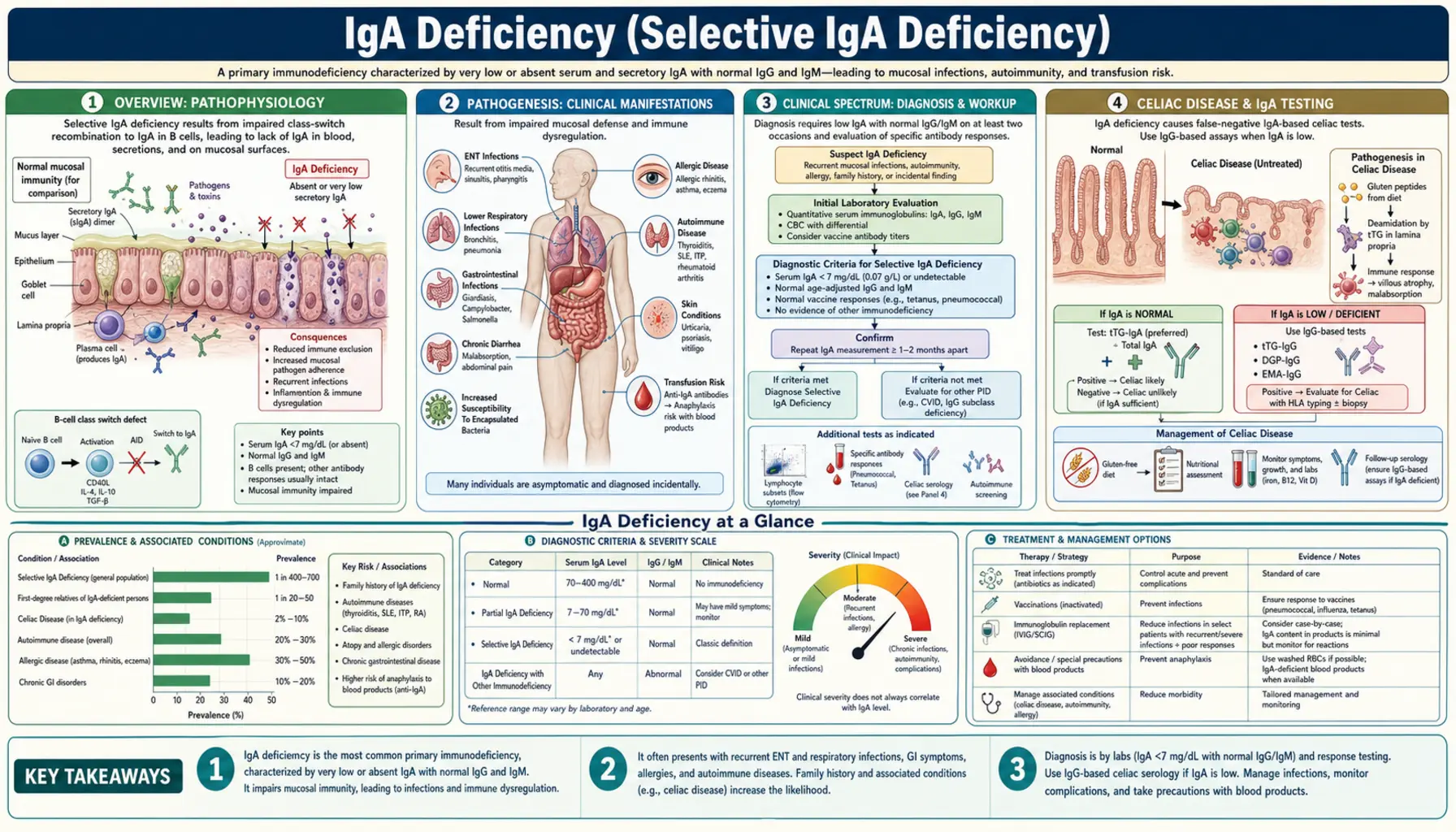
Selective IgA deficiency (SIgAD) is the most common primary immunodeficiency worldwide, affecting approximately 1 in 300–500 individuals of Caucasian descent and 1 in 16,000 Japanese individuals — a striking ethnic variation that points to underlying genetic architecture. It is defined as a serum IgA level below 7 mg/dL with normal serum IgG and IgM in patients aged 4 years or older, after excluding secondary causes.
IgA is the principal secretory immunoglobulin of the body's mucosal surfaces. Secretory IgA (sIgA) is the dominant antibody in saliva, tears, colostrum, breast milk, intestinal secretions, and the respiratory mucosa — the very surfaces that act as the body's first-line barrier against pathogens. Despite being the most abundant antibody class produced each day (approximately 3–5 grams secreted daily across all mucosal surfaces combined), its absence is remarkably well tolerated in many individuals because serum IgG and IgM can partially compensate.
Partial IgA deficiency is defined as serum IgA between 7 mg/dL and 70 mg/dL with normal IgG and IgM. True selective IgA deficiency requires IgA below 7 mg/dL with normal IgG and IgM. When IgG is also low, the diagnosis shifts toward common variable immunodeficiency (CVID), a more serious disorder that shares genetic overlap with SIgAD.
Pathogenesis
The central defect in selective IgA deficiency is a failure of B cell terminal differentiation. IgA-committed B cells are present and circulate in normal numbers, but they cannot complete the class-switch recombination to IgA or fully mature into IgA-secreting plasma cells. The block occurs late in B cell development — beyond the common lymphoid progenitor and class-switch machinery — making SIgAD distinct from agammaglobulinemias, where B cells are absent entirely.
Genetic associations: The strongest HLA association is the A1-B8-DR3 haplotype, particularly common in Northern Europeans — explaining the high prevalence in Caucasian populations. Chromosomal deletions at 22q11.2 and 18q are identified in a subset of patients, especially those with additional dysmorphic features. Mutations in TACI (TNFRSF13B), encoding a B cell receptor for BAFF and APRIL, are found in a minority of both SIgAD and CVID patients, reinforcing the shared genetic substrate of these two conditions. Familial clustering occurs, and first-degree relatives of CVID patients have elevated rates of SIgAD.
Secondary (drug-induced) IgA deficiency is clinically important because it is reversible. Implicated agents include phenytoin, valproate, sulfasalazine, captopril, D-penicillamine, and gold salts. The mechanism is thought to involve interference with cytokine signaling required for IgA class switching. Discontinuing the offending drug typically leads to normalization of IgA levels over months.
Clinical Spectrum
One of the most striking features of selective IgA deficiency is its clinical heterogeneity. The majority of affected individuals — estimates range from 50% to 85% — are entirely asymptomatic and are discovered incidentally when serum protein electrophoresis or immunoglobulin panels are ordered for unrelated reasons. For these individuals, the diagnosis may carry little practical consequence beyond the blood product risk described below.
Recurrent sinopulmonary infections are the most common symptomatic manifestation. Organisms that colonize mucosal surfaces — Streptococcus pneumoniae and Haemophilus influenzae — are the usual culprits. Affected individuals may have recurrent sinusitis, chronic otitis media, and pneumonia, sometimes progressing to bronchiectasis with repeated pulmonary infections over years.
Gastrointestinal manifestations deserve particular attention. Giardia lamblia is normally cleared from the small intestine by secretory IgA; IgA-deficient patients are highly susceptible to giardiasis and may suffer protracted, recurrent infections that are difficult to eradicate. Celiac disease occurs 10 times more often in IgA-deficient patients than in the general population (discussed separately below). Inflammatory bowel disease and nodular lymphoid hyperplasia of the small bowel are additional GI associations.
Autoimmune diseases are found in 20–30% of symptomatic patients. Systemic lupus erythematosus (SLE) is present in 10–15% of IgA-deficient individuals. Rheumatoid arthritis, autoimmune thyroid diseases (both Hashimoto's thyroiditis and Graves' disease), vitiligo, type 1 diabetes mellitus, and autoimmune hepatitis are all overrepresented. The mechanism likely involves dysregulation of mucosal immune tolerance — the absence of secretory IgA disrupts the normal suppression of immune responses to commensal antigens, tilting the balance toward autoimmunity.
Atopy is paradoxically increased. Asthma, allergic rhinitis, atopic dermatitis, and food allergies are more common in SIgAD patients, possibly because the absence of mucosal IgA permits allergen penetration and aberrant Th2 sensitization at epithelial surfaces.
Celiac Disease and IgA Testing
The intersection of IgA deficiency and celiac disease creates a critical diagnostic trap that every clinician ordering celiac antibody tests must understand. Celiac disease occurs approximately 10 times more frequently in patients with selective IgA deficiency than in the general population — making the co-occurrence far from rare. Yet the standard celiac screening tests are IgA-based antibodies: anti-tissue transglutaminase IgA (anti-tTG IgA) and anti-endomysial IgA (EMA-IgA). In a patient with IgA deficiency, both of these tests will be falsely negative, even in the presence of active celiac disease with severe villous atrophy.
This false-negative result is not a testing error — it is a predictable consequence of the underlying immunodeficiency. When the immune system cannot produce IgA, it cannot produce IgA antibodies against tissue transglutaminase, regardless of how much gluten damage is occurring in the small intestine. A patient with unrecognized celiac disease and IgA deficiency may receive a falsely reassuring celiac screen and go undiagnosed for years.
The solution is straightforward but requires awareness: always check total serum IgA when ordering celiac antibody panels. If serum IgA is low or undetectable, use IgG-based celiac antibodies instead — anti-tTG IgG and anti-deamidated gliadin peptide IgG (anti-DGP IgG). These tests retain their sensitivity for celiac disease even in IgA-deficient patients. Small bowel biopsy (Marsh grading via duodenal histology) remains the gold standard and does not depend on immunoglobulin class.
Many major laboratory test requisitions now automatically reflexively report total IgA when anti-tTG IgA is ordered. Clinicians should verify that their laboratory follows this practice and interpret any IgA-based celiac antibody result in the context of the patient's total serum IgA level.
Anti-IgA Antibodies and Blood Product Risk
Approximately 40% of patients with complete IgA deficiency develop IgE-class anti-IgA antibodies. These antibodies are directed against IgA determinants that the patient's immune system has never seen — treating exogenous IgA as foreign. When such a patient is exposed to IgA-containing blood products, these pre-formed IgE anti-IgA antibodies can trigger immediate hypersensitivity reactions ranging from urticaria and flushing to severe, life-threatening anaphylaxis.
Blood products that contain IgA include: whole blood, packed red blood cells, fresh frozen plasma, platelet concentrates, and most standard intravenous immunoglobulin (IVIG) preparations. Even trace amounts of IgA in washed red blood cells or standard IVIG can be sufficient to trigger anaphylaxis in highly sensitized patients.
Safe management strategies:
- Test for anti-IgA antibodies in any IgA-deficient patient who requires blood products or IVIG. High-titer IgE anti-IgA antibodies confer the greatest anaphylaxis risk.
- Use IgA-depleted IVIG if IVIG is truly indicated. Products with IgA content below 1 µg/mL (e.g., Gammagard Liquid) are specifically formulated for IgA-deficient patients. Standard IVIG preparations typically contain 720 µg/mL or more of IgA.
- Use washed red blood cells for transfusions — repeated washing removes most IgA from donor erythrocyte preparations.
- Document anti-IgA status prominently in the medical record and ensure the patient carries alert documentation (medical alert bracelet, wallet card).
- Have anaphylaxis treatment immediately available (epinephrine, antihistamines, corticosteroids) for any blood product administration.
- Pre-medication with antihistamines and corticosteroids can reduce but does not eliminate the risk of transfusion reactions.
It is worth noting that routine IVIG therapy is generally not indicated for uncomplicated SIgAD — secretory IgA cannot be replaced by intravenous infusion, and the anaphylaxis risk outweighs the benefit for most patients. IVIG is reserved for the subset who develop CVID-like hypogammaglobulinemia or severe recurrent infections despite other measures.
Evolution to CVID
Selective IgA deficiency and common variable immunodeficiency (CVID) are not entirely separate diseases — they exist on a continuum of B cell maturation failure, sharing genetic underpinnings and clinical overlap. Approximately 15–20% of patients with SIgAD will eventually develop CVID over the course of years to decades. This transition is defined by a progressive decline in serum IgG (and sometimes IgM) to levels below normal, accompanied by increasing susceptibility to bacterial infections.
Monitoring: Because of this progression risk, annual measurement of serum immunoglobulin levels (IgG, IgA, IgM) is recommended for all patients with SIgAD, particularly those who are symptomatic or who have relatives with CVID. A falling IgG trajectory — even within the low-normal range — should prompt more frequent monitoring and consideration of prophylactic antibiotics or IVIG replacement.
Shared genetic architecture: TACI mutations (TNFRSF13B) are found in both conditions, and families have been described where some members have SIgAD and others have CVID — suggesting that additional genetic modifiers determine whether the defect remains limited to IgA or expands to affect IgG and IgM production. HLA associations (particularly HLA-DR3 and HLA-B8) are shared across both conditions in European populations.
Practical implication: a diagnosis of SIgAD should not be viewed as a static, benign finding. It is a marker of B cell immune dysfunction that warrants long-term follow-up, not a one-time laboratory note and discharge.
Diagnosis
The diagnosis of selective IgA deficiency is established by measuring serum immunoglobulin levels. The key criteria are:
- Serum IgA below 7 mg/dL on at least two separate measurements.
- Age 4 years or older — physiologically low IgA in infants and young children (under 4) does not constitute SIgAD; many infants have transient hypogammaglobulinemia that resolves spontaneously.
- Normal serum IgG and IgM — if IgG is also significantly reduced, CVID must be considered and evaluated.
- Exclusion of secondary causes — review all medications (phenytoin, valproate, sulfasalazine, captopril, D-penicillamine, gold) before attributing IgA deficiency to a primary immunodeficiency.
Additional diagnostic workup:
- Anti-IgA antibody testing — essential before any blood product administration. Identifies patients at anaphylaxis risk.
- Celiac screen with IgG-based antibodies — anti-tTG IgG and anti-DGP IgG. Never rely on IgA-based tests in a confirmed IgA-deficient patient.
- Autoimmune panel — ANA, anti-dsDNA, thyroid antibodies (anti-TPO, anti-thyroglobulin), rheumatoid factor as clinically indicated.
- Functional antibody responses — post-vaccination titers (pneumococcal, tetanus) assess whether IgG responses are adequate, helping stratify infection risk.
- Genetic testing — TACI/TNFRSF13B sequencing in familial cases or when CVID is also suspected in family members.
- Annual IgG monitoring — baseline and annual serum IgG to detect early CVID evolution.
Measurement method is nephelometry (the standard for clinical immunoglobulin quantification). False low values can occur with paraprotein interference; if clinical suspicion is high despite low nephelometric IgA, protein electrophoresis with immunofixation can clarify.
Treatment
There is no therapy that restores IgA production in selective IgA deficiency. Management is therefore focused on preventing complications, treating infections promptly, monitoring for disease evolution, and avoiding iatrogenic harm from blood product exposure.
Asymptomatic SIgAD: No pharmacological treatment is needed. Counsel the patient about:
- The anti-IgA antibody risk and blood product precautions.
- The importance of informing any anesthesiologist, surgeon, or blood bank before procedures.
- Annual serum IgG monitoring for CVID evolution.
- The need for IgG-based celiac antibody testing if celiac disease is ever suspected.
Recurrent sinopulmonary infections: Prophylactic antibiotics (e.g., azithromycin, trimethoprim-sulfamethoxazole) are appropriate for patients with two or more serious infections per year. Pneumococcal and influenza vaccination is strongly recommended — vaccine responses are generally adequate (IgG-mediated) despite IgA deficiency. Early and aggressive antibiotic treatment of acute infections prevents bronchiectasis.
IVIG therapy: Standard IVIG is generally contraindicated in SIgAD due to anaphylaxis risk from IgA content. If replacement immunoglobulin is truly necessary (e.g., for evolution toward CVID with falling IgG), use only IgA-depleted IVIG preparations (IgA content <1 µg/mL). Start at low doses with close monitoring in a facility equipped to manage anaphylaxis.
Celiac disease: A strict, lifelong gluten-free diet. Nutritional monitoring (iron, folate, vitamin B12, vitamin D, calcium) for malabsorption. Bone density monitoring (DEXA scan) given celiac-related osteoporosis risk.
Autoimmune manifestations: Treat according to the specific autoimmune diagnosis (e.g., levothyroxine for hypothyroidism, hydroxychloroquine and sun protection for SLE, disease-modifying antirheumatic drugs for RA). The underlying IgA deficiency does not typically alter the treatment of the autoimmune condition itself, though some disease-modifying drugs (sulfasalazine, gold, D-penicillamine) can worsen IgA deficiency and should be used cautiously.
Giardiasis: Metronidazole or tinidazole. Recurrent infections may require prolonged treatment or repeated courses. Avoid institutional water sources in endemic areas.
Key Research Papers
The following peer-reviewed studies establish the foundational evidence base for selective IgA deficiency — its epidemiology, pathogenesis, clinical associations, and management.
- Hammarström L, et al. (2000). Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clinical and Experimental ImmunologySearch PubMed
- Yel L. (2010). Selective IgA deficiency. Journal of Clinical ImmunologySearch PubMed
- Cunningham-Rundles C. (2001). Physiology of IgA and IgA deficiency. Journal of Clinical ImmunologySearch PubMed
- Cataldo F, et al. (1998). IgA antiendomysium antibodies in identifying occult coeliac disease in children with type 1 diabetes mellitus. GutSearch PubMed
- Kowalczyk D, et al. (1994). Anti-IgA antibodies in selective IgA deficiency and in various immunodeficiency disorders. Clinical and Experimental ImmunologySearch PubMed
- Liblau RS, Bach JF. (1992). Selective IgA deficiency and autoimmunity. International Archives of Allergy and ImmunologySearch PubMed
- Abolhassani H, et al. (2018). Worldwide variable immunodeficiency registry: report on the global distribution of primary immunodeficiency diseases. Journal of Clinical ImmunologySearch PubMed
- Sekine H, et al. (2007). Role of TACI in CVID and IgA deficiency. Journal of Clinical ImmunologySearch PubMed
- Aghamohammadi A, et al. (2009). Progression of selective IgA deficiency to common variable immunodeficiency. International Archives of Allergy and ImmunologySearch PubMed
- Notarangelo L, et al. (2009). Primary immunodeficiencies: 2009 update. Journal of Allergy and Clinical ImmunologySearch PubMed
- Wang N, et al. (2003). Heterozygous mutations in the gene encoding TACI are associated with common variable immunodeficiency and selective IgA deficiency. Nature GeneticsSearch PubMed
- Schaffer FM, et al. (1991). Individuals with IgA deficiency and common variable immunodeficiency share polymorphisms of major histocompatibility complex class III genes. Proceedings of the National Academy of SciencesSearch PubMed
PubMed Topic Searches:
- Selective IgA deficiency — PubMed
- IgA deficiency + celiac disease — PubMed
- Anti-IgA antibodies and anaphylaxis — PubMed
- IgA deficiency to CVID progression — PubMed
Connections
- Immunology
- Common Variable Immunodeficiency (CVID)
- Severe Combined Immunodeficiency (SCID)
- X-Linked Agammaglobulinemia
- Wiskott-Aldrich Syndrome
- Chronic Granulomatous Disease
- Celiac Disease
- All Conditions